Recombinant Strains of Human Parechovirus in Rural Areas in the North of Brazil

 ,
, 
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Sample Processing
2.3. Alignment and Phylogenetic Analysis
2.4. Detection of Recombination
2.5. Coalescent Analysis
3. Results
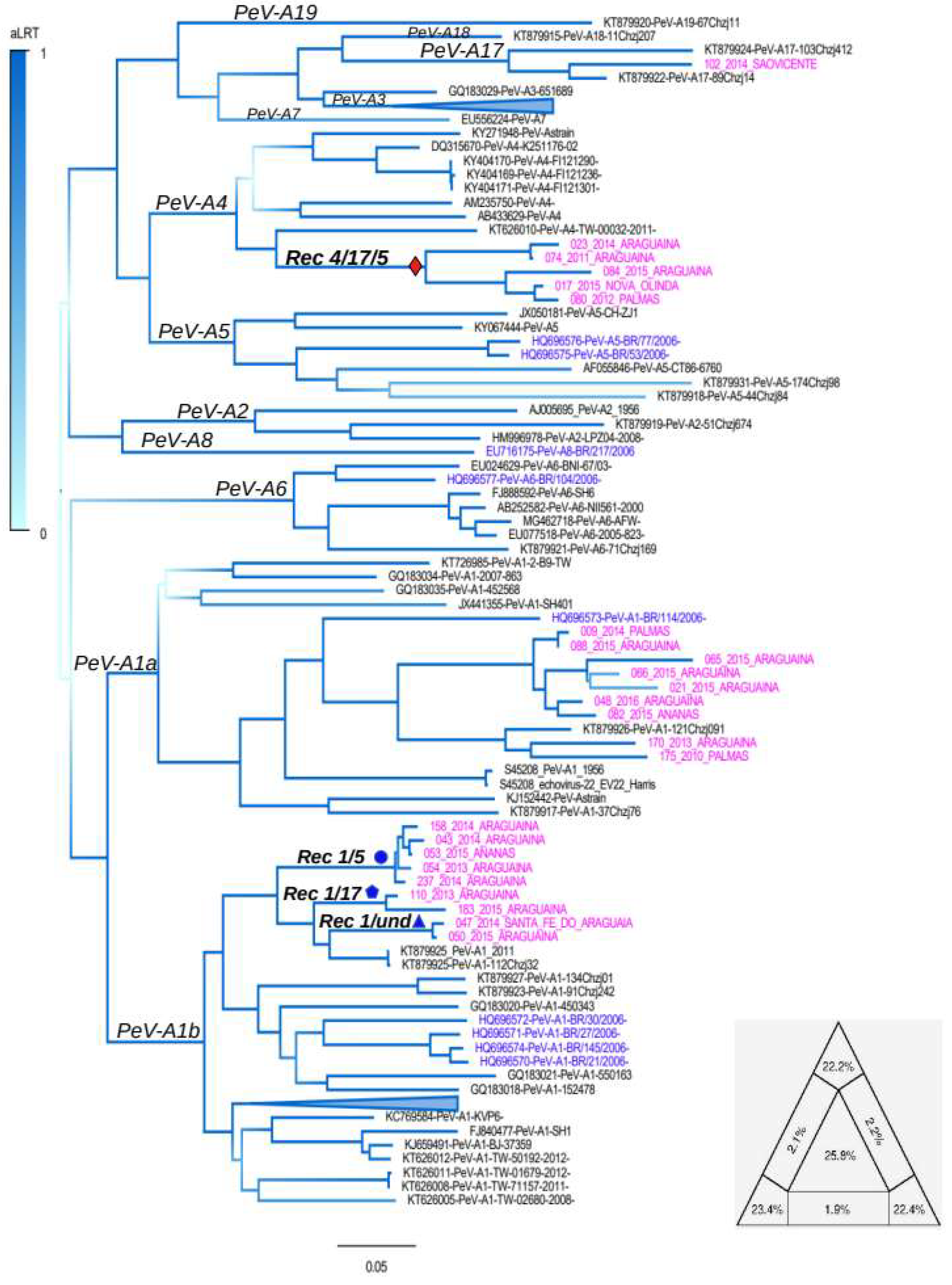
3.1. Genotyping Tree
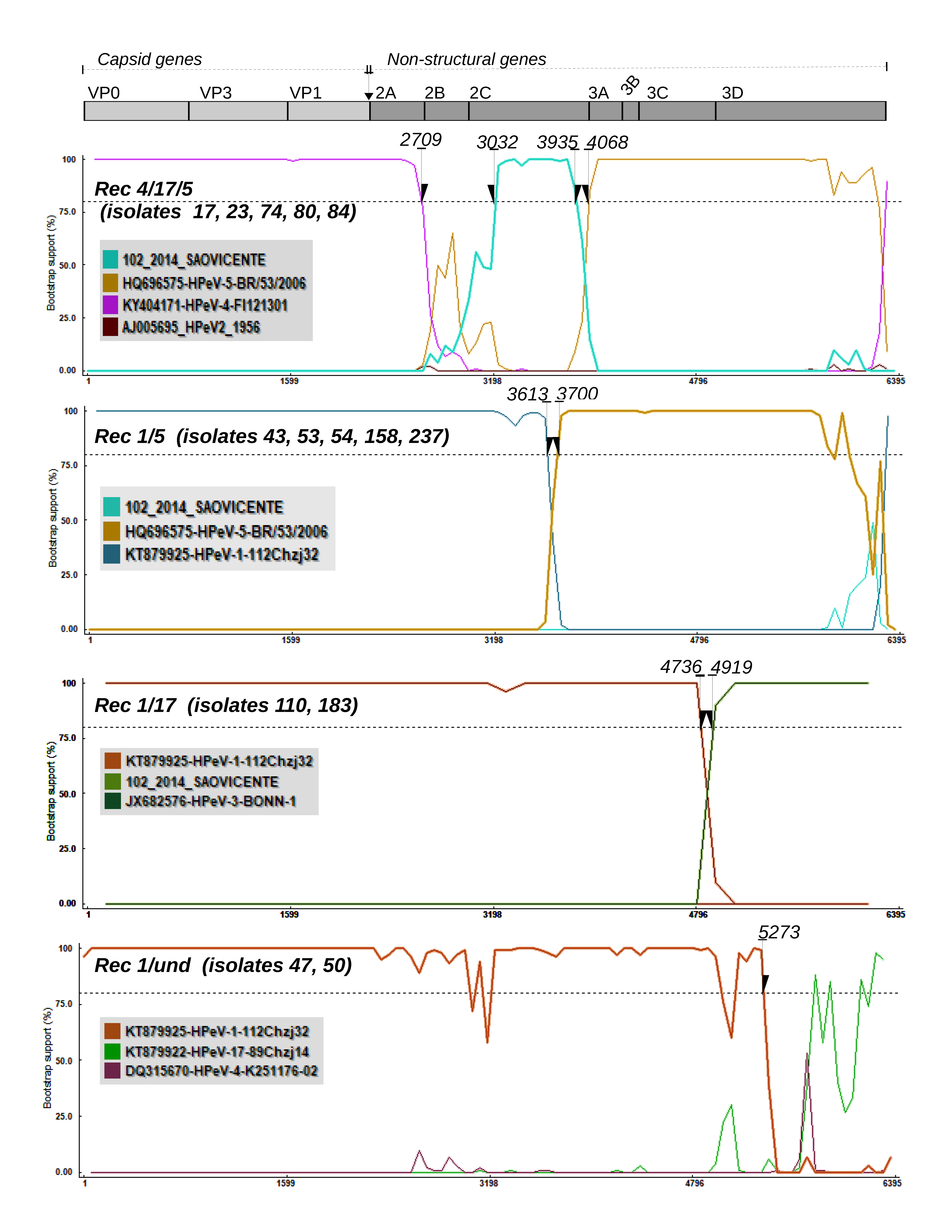
3.2. Recombination Analysis
3.3. Mosaic Pattern
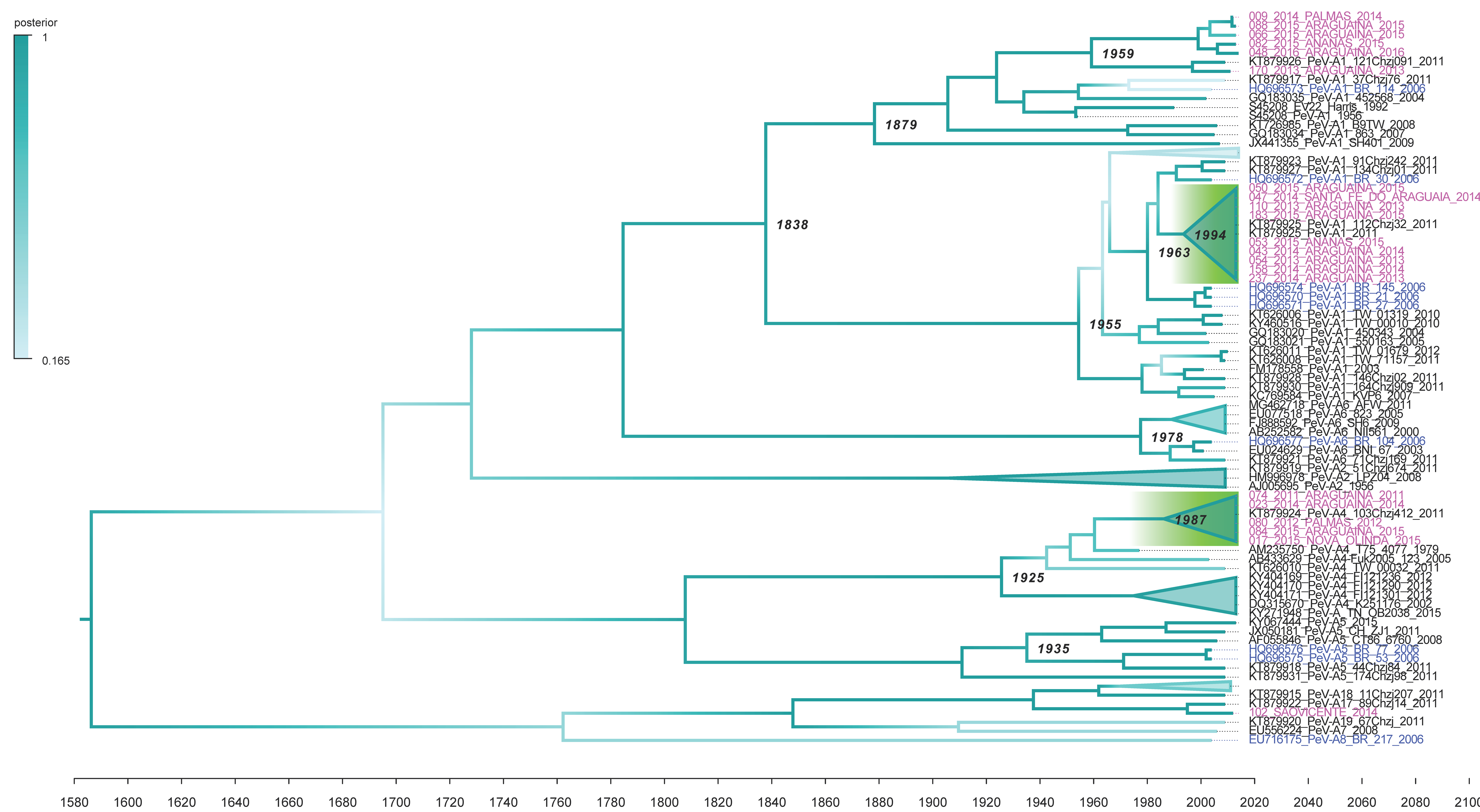
3.4. Dynamics of PeV-A Infection
3.5. Timescale for the PeV-A Recombinant Strains in Brazil
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV virus taxonomy profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Galama, J.M.; van Kuppeveld, F.J. Identification of potential recombination breakpoints in human parechoviruses. J. Virol. 2009, 83, 3379–3383. [Google Scholar] [CrossRef]
- Olijve, L.; Jennings, L.; Walls, T. Human parechovirus: An increasingly recognized cause of sepsis-like illness in young infants. Clin. Microbiol. Rev. 2018, 31, e00047-17. [Google Scholar] [CrossRef]
- Hyypia, T.; Horsnell, C.; Maaronen, M.; Khan, M.; Kalkkinen, N.; Auvinen, P.; Kinnunen, L.; Stanway, G. A distinct picornavirus group identified by sequence analysis. Proc. Natl. Acad. Sci. USA 1992, 89, 8847–8851. [Google Scholar] [CrossRef]
- Ito, M.; Yamashita, T.; Tsuzuki, H.; Takeda, N.; Sakae, K. Isolation and identification of a novel human parechovirus. J. Gen. Virol. 2004, 85, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Stanway, G.; Kalkkinen, N.; Roivainen, M.; Ghazi, F.; Khan, M.; Smyth, M.; Meurman, O.; Hyypia, T. Molecular and biological characteristics of echovirus 22, a representative of a new picornavirus group. J. Virol. 1994, 68, 8232–8238. [Google Scholar] [PubMed]
- Aizawa, Y.; Izumita, R.; Saitoh, A. Human parechovirus type 3 infection: An emerging infection in neonates and young infants. J. Infect. Chemother. 2017, 23, 419–426. [Google Scholar] [CrossRef]
- Zhang, D.L.; Jin, Y.; Li, D.D.; Cheng, W.X.; Xu, Z.Q.; Yu, J.M.; Jin, M.; Yang, S.H.; Zhang, Q.; Cui, S.X.; et al. Prevalence of human parechovirus in chinese children hospitalized for acute gastroenteritis. Clin. Microbiol. Infect. 2011, 17, 1563–1569. [Google Scholar] [CrossRef]
- Esposito, S.; Rahamat-Langendoen, J.; Ascolese, B.; Senatore, L.; Castellazzi, L.; Niesters, H.G. Pediatric parechovirus infections. J. Clin. Virol. 2014, 60, 84–89. [Google Scholar] [CrossRef]
- Chiang, G.P.K.; Chen, Z.; Chan, M.C.W.; Lee, S.H.M.; Kwok, A.K.; Yeung, A.C.M.; Nelson, E.A.S.; Hon, K.L.; Leung, T.F.; Chan, P.K.S. Clinical features and seasonality of parechovirus infection in an Asian subtropical city, Hong Kong. PLoS ONE 2017, 12, e0184533. [Google Scholar] [CrossRef] [PubMed]
- Chuchaona, W.; Khamrin, P.; Yodmeeklin, A.; Saikruang, W.; Kongsricharoern, T.; Ukarapol, N.; Okitsu, S.; Hayakawa, S.; Ushijima, H.; Maneekarn, N. Detection and characterization of a novel human parechovirus genotype in Thailand. Infect. Genet. Evol. 2015, 31, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Harvala, H.; Wolthers, K.C.; Simmonds, P. Parechoviruses in children: Understanding a new infection. Curr. Opin. Infect. Dis. 2010, 23, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.H.; Panayiotou, M.; Girling, G.D.; Peard, C.I.; Oikarinen, S.; Hyoty, H.; Stanway, G. Evolution and conservation in human parechovirus genomes. J. Gen. Virol. 2009, 90, 1702–1712. [Google Scholar] [CrossRef]
- Benschop, K.S.; Williams, C.H.; Wolthers, K.C.; Stanway, G.; Simmonds, P. Widespread recombination within human parechoviruses: Analysis of temporal dynamics and constraints. J. Gen. Virol. 2008, 89, 1030–1035. [Google Scholar] [CrossRef]
- Drexler, J.F.; Grywna, K.; Lukashev, A.; Stocker, A.; Almeida, P.S.; Wieseler, J.; Ribeiro, T.C.; Petersen, N.; Ribeiro Hda, C., Jr.; Belalov, I.; et al. Full genome sequence analysis of parechoviruses from Brazil reveals geographical patterns in the evolution of non-structural genes and intratypic recombination in the capsid region. J. Gen. Virol. 2011, 92, 564–571. [Google Scholar] [CrossRef]
- da Costa, A.C.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; das Chagas, R.T.; Abrao, M.; Soares, C.; et al. Recombination located over 2a-2b junction ribosome frameshifting region of saffold cardiovirus. Viruses 2018, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.C.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; das Chagas, R.T.; Abrao, M.; Soares, C.; et al. Near full length genome of a recombinant (e/d) cosavirus strain from a rural area in the central region of Brazil. Sci. Rep. 2018, 8, 12304. [Google Scholar] [CrossRef]
- Charlys da Costa, A.; Theze, J.; Komninakis, S.C.V.; Sanz-Duro, R.L.; Felinto, M.R.L.; Moura, L.C.C.; Barroso, I.M.O.; Santos, L.E.C.; Nunes, M.A.L.; Moura, A.A.; et al. Spread of chikungunya virus east/central/south African genotype in northeast Brazil. Emerg. Infect. Dis. 2017, 23, 1742–1744. [Google Scholar] [CrossRef]
- Li, L.; Deng, X.; Mee, E.T.; Collot-Teixeira, S.; Anderson, R.; Schepelmann, S.; Minor, P.D.; Delwart, E. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J. Virol. Methods 2015, 213, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, É.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Posada, D. Jmodeltest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. Tree-puzzle: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Suchard, M.A. Genealogical working distributions for bayesian model testing with phylogenetic uncertainty. Syst. Biol. 2016, 65, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; de Vries, M.; van Hemert, F.J.; Benschop, K.; van der Hoek, L. Rooting human parechovirus evolution in time. BMC Evol. Biol. 2009, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Villanova, F.; Cui, S.; Ai, X.; Leal, E. Analysis of full-length genomes of porcine teschovirus (PTV) and the effect of purifying selection on phylogenetic trees. Arch. Virol. 2016, 161, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Branch-length prior influences bayesian posterior probability of phylogeny. Syst. Biol. 2005, 54, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using beast 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Grywna, K.; Stocker, A.; Almeida, P.S.; Medrado-Ribeiro, T.C.; Eschbach-Bludau, M.; Petersen, N.; da Costa-Ribeiro, H., Jr.; Drosten, C. Novel human parechovirus from Brazil. Emerg. Infect. Dis. 2009, 15, 310–313. [Google Scholar] [CrossRef]
- Calvert, J.; Chieochansin, T.; Benschop, K.S.; McWilliam Leitch, E.C.; Drexler, J.F.; Grywna, K.; da Costa Ribeiro, H., Jr.; Drosten, C.; Harvala, H.; Poovorawan, Y.; et al. Recombination dynamics of human parechoviruses: Investigation of type-specific differences in frequency and epidemiological correlates. J. Gen. Virol. 2010, 91, 1229–1238. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vp1 gene (n = 122, l = 556) | |||
|---|---|---|---|
| Coalescent Model | MLE | Substitution Rates * | tMRCA * |
| Constant size | −16,618.29 | 2.2 × 10−3(1.6 × 10−3–2.7 × 10−3) | 1552 (1270–1760) |
| Exponential growth | −16,696.69 | 1.8 × 10−3(1.1 × 10–2.5 × 10−3) | 1489 (1143–1747) |
| BSL | −17,212.22 | 1.7 × 10−3(1.0 × 10−3–2.5 × 10−3) | 1482 (1248–1780) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, É.; Luchs, A.; Milagres, F.A.d.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.C.A.B.S.; Brustulin, R.; Chagas, R.T.d.; Abrão, M.d.F.N.d.S.; Soares, C.V.d.D.A.; et al. Recombinant Strains of Human Parechovirus in Rural Areas in the North of Brazil. Viruses 2019, 11, 488. https://doi.org/10.3390/v11060488
Leal É, Luchs A, Milagres FAdP, Komninakis SV, Gill DE, Lobato MCABS, Brustulin R, Chagas RTd, Abrão MdFNdS, Soares CVdDA, et al. Recombinant Strains of Human Parechovirus in Rural Areas in the North of Brazil. Viruses. 2019; 11(6):488. https://doi.org/10.3390/v11060488
Chicago/Turabian StyleLeal, Élcio, Adriana Luchs, Flávio Augusto de Pádua Milagres, Shirley Vasconcelos Komninakis, Danielle Elise Gill, Márcia Cristina Alves Brito Sayão Lobato, Rafael Brustulin, Rogério Togisaki das Chagas, Maria de Fátima Neves dos Santos Abrão, Cássia Vitória de Deus Alves Soares, and et al. 2019. "Recombinant Strains of Human Parechovirus in Rural Areas in the North of Brazil" Viruses 11, no. 6: 488. https://doi.org/10.3390/v11060488
APA StyleLeal, É., Luchs, A., Milagres, F. A. d. P., Komninakis, S. V., Gill, D. E., Lobato, M. C. A. B. S., Brustulin, R., Chagas, R. T. d., Abrão, M. d. F. N. d. S., Soares, C. V. d. D. A., Villanova, F., Witkin, S. S., Deng, X., Sabino, E. C., Delwart, E., & Costa, A. C. d. (2019). Recombinant Strains of Human Parechovirus in Rural Areas in the North of Brazil. Viruses, 11(6), 488. https://doi.org/10.3390/v11060488