Classifying the Unclassified: A Phage Classification Method


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources
2.1.1. Phages
2.1.2. Host Genomes
2.2. Data Preparation
2.3. Profile HMM Construction
2.4. Profile HMM Refinement
2.5. CDS Prediction and Additional HMM Refinement
2.6. Software Tools
3. Results and Discussion
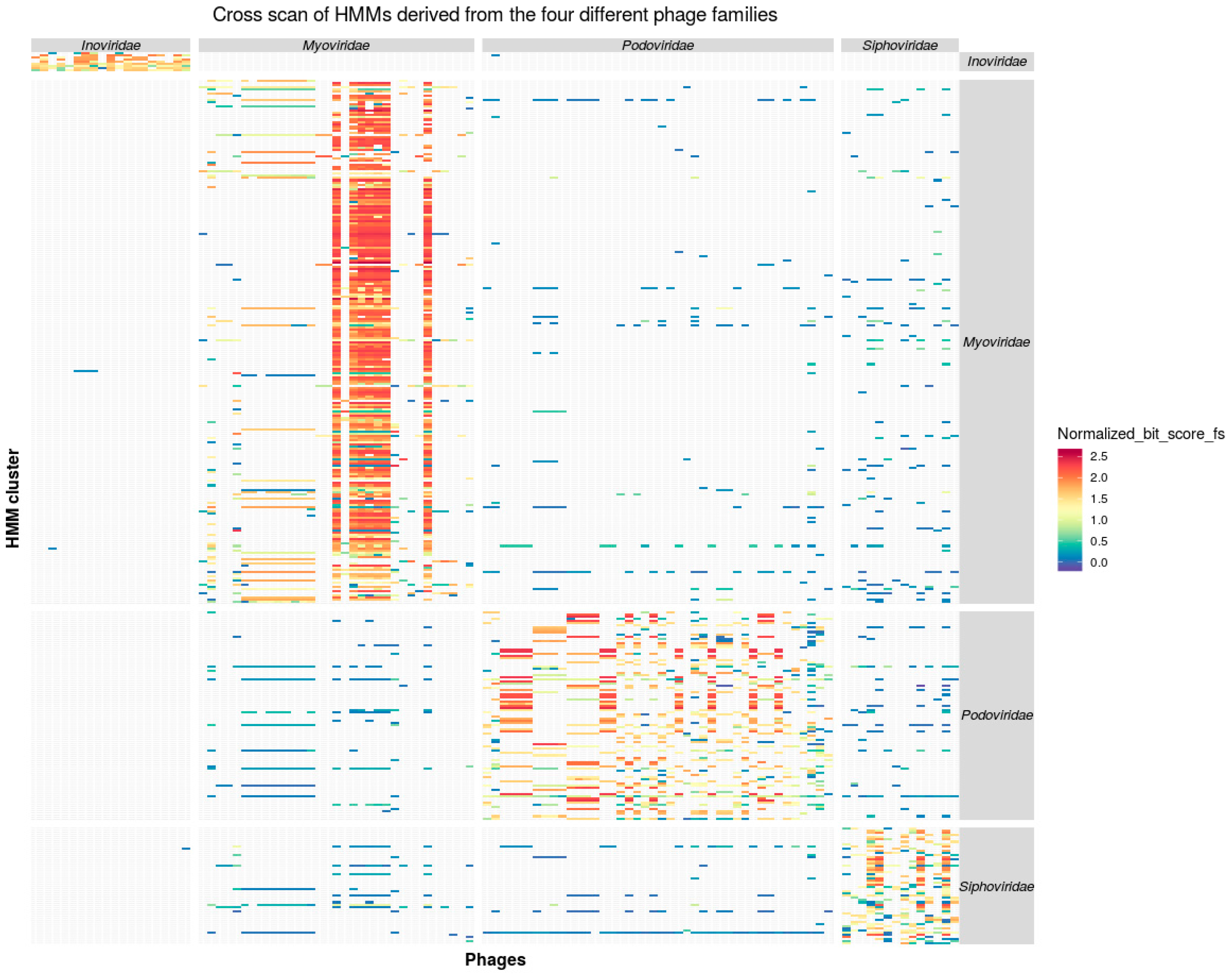
3.1. Phage Protein Families and Profile HMMs
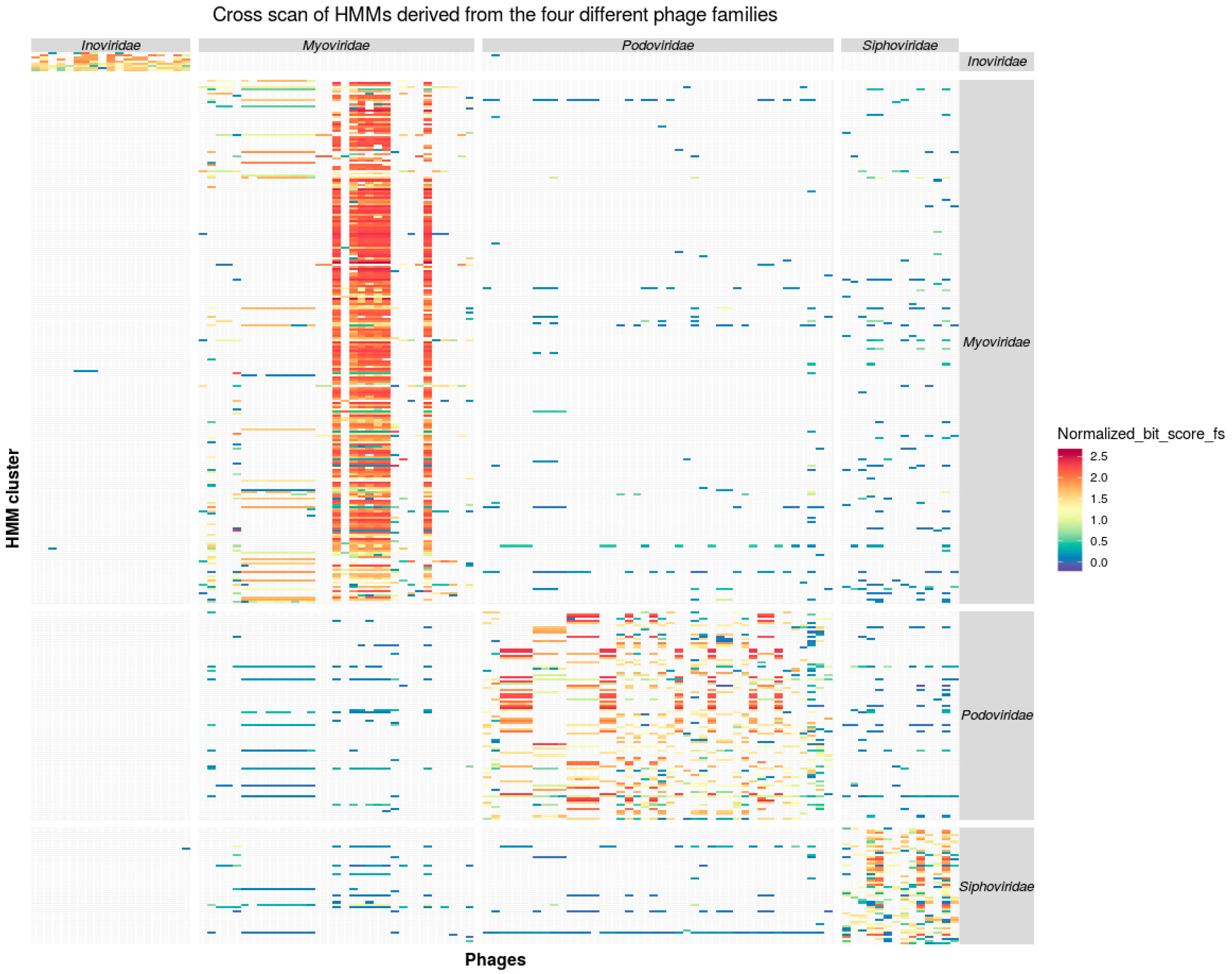
3.2. Taxon Specificity of the Protein Family Models
3.3. Consistency of Taxon-Specific HMMs
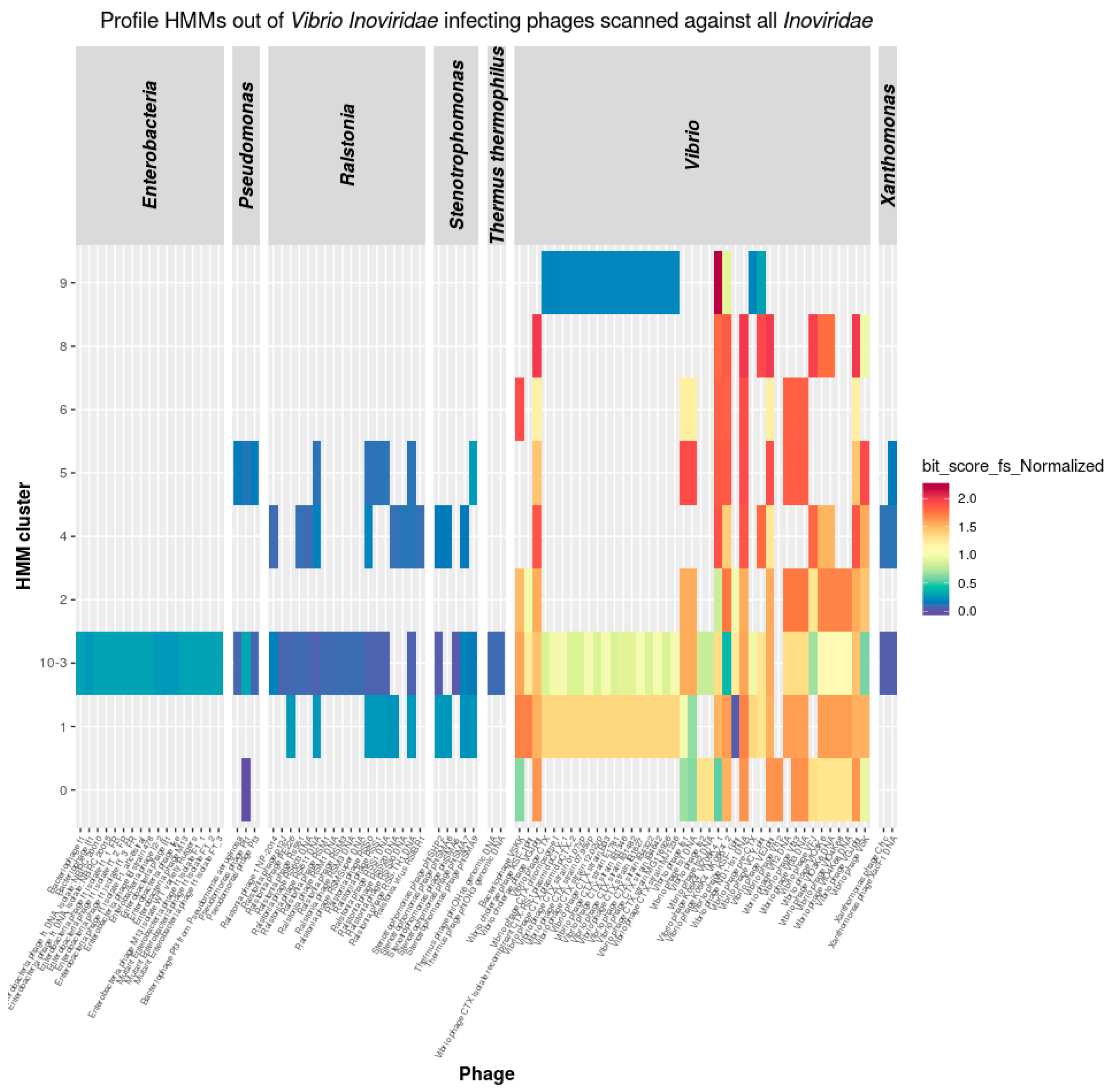
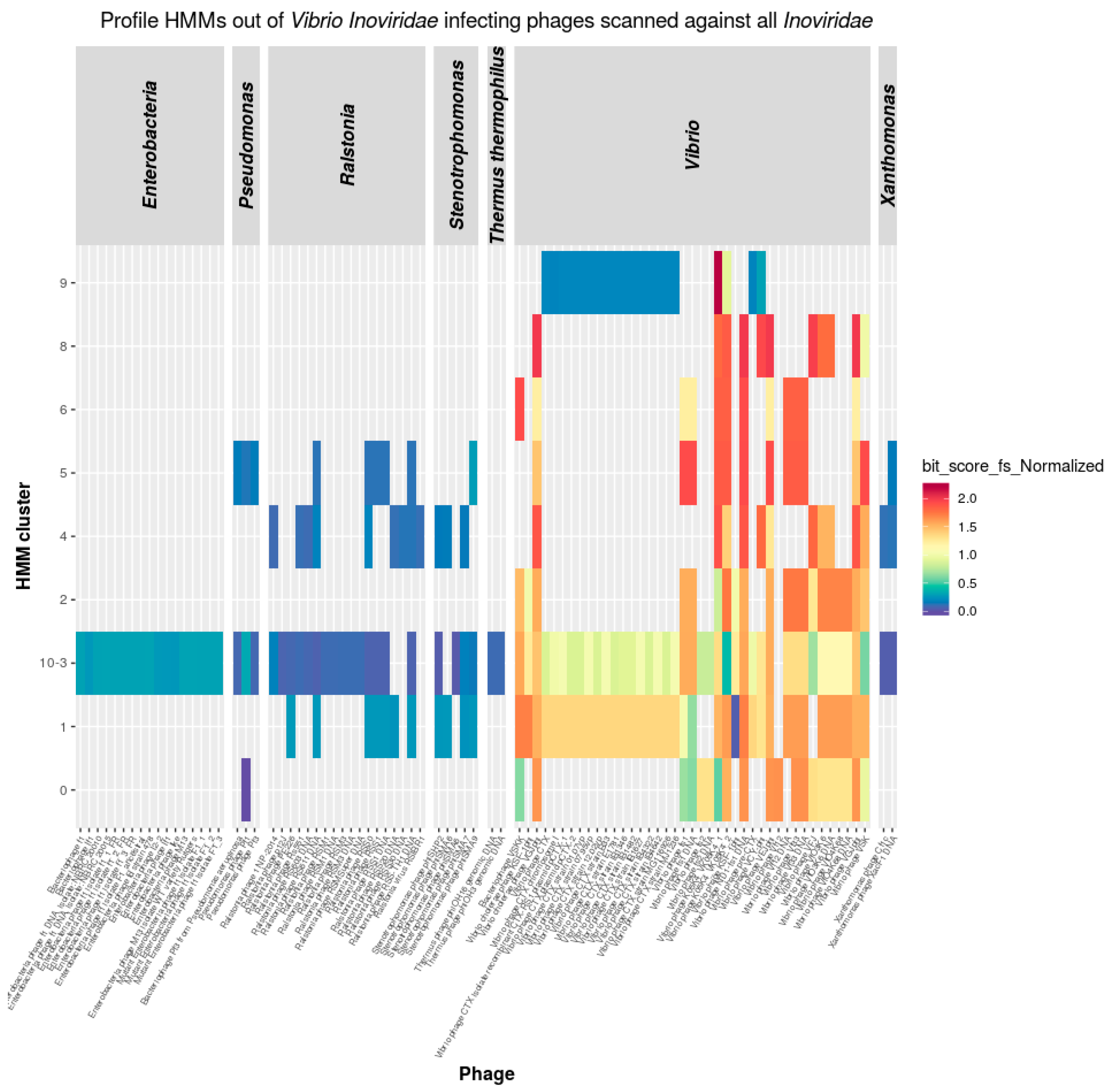
3.3.1. Inoviridae
3.3.2. Caudovirales
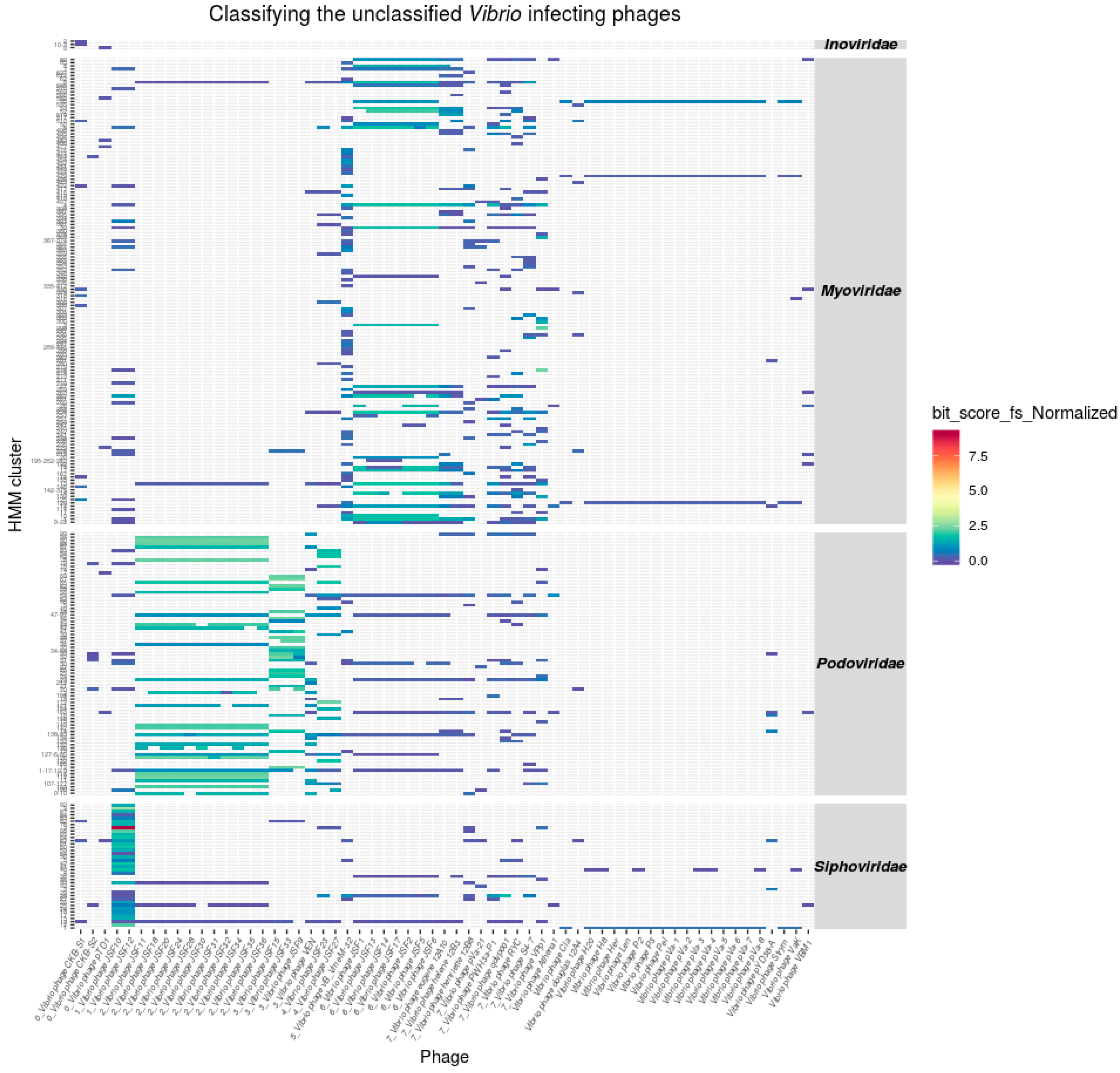
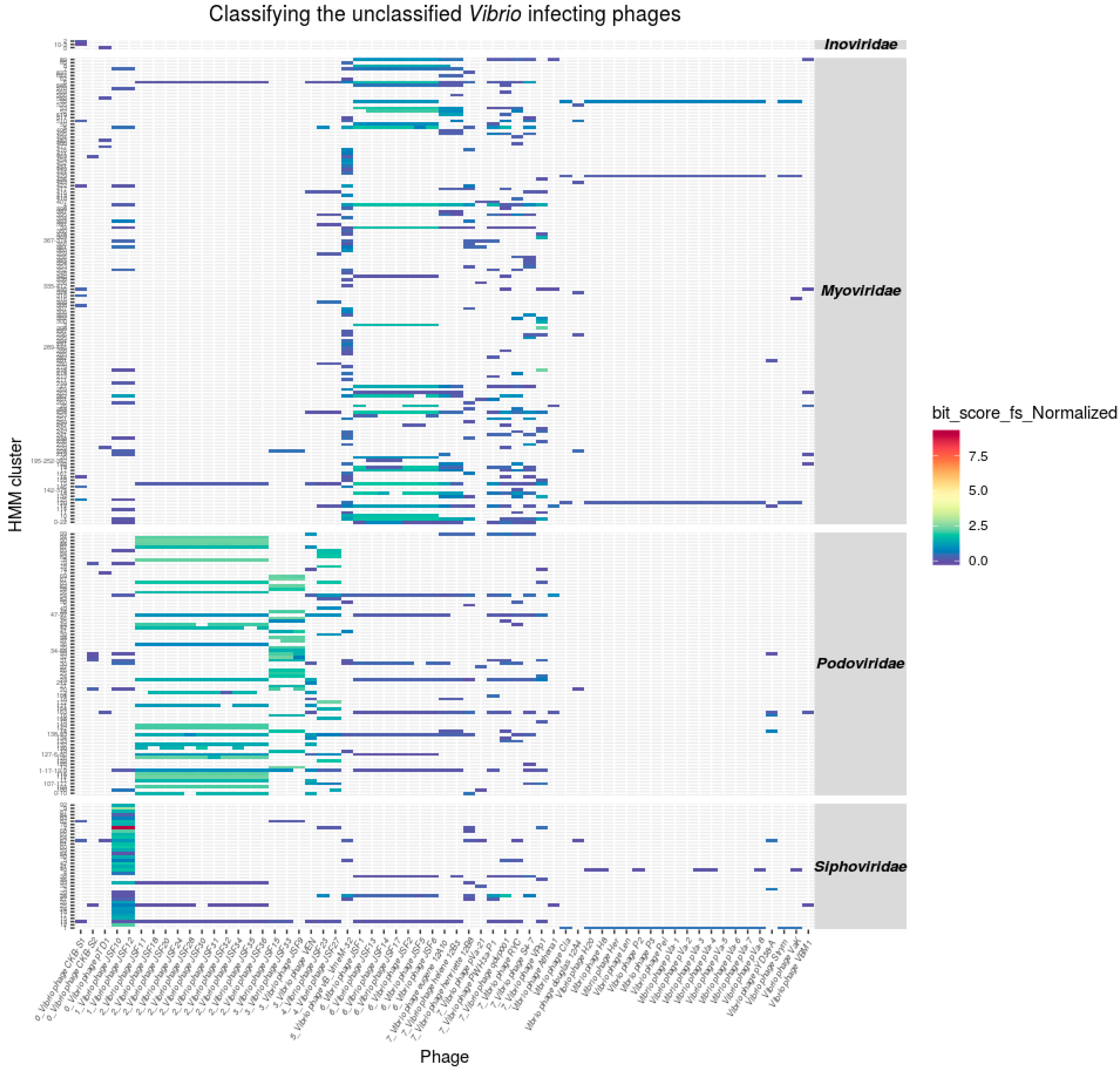
3.4. Classification of Unclassified Phages
Taxonomic Assignments of Tailed Phages
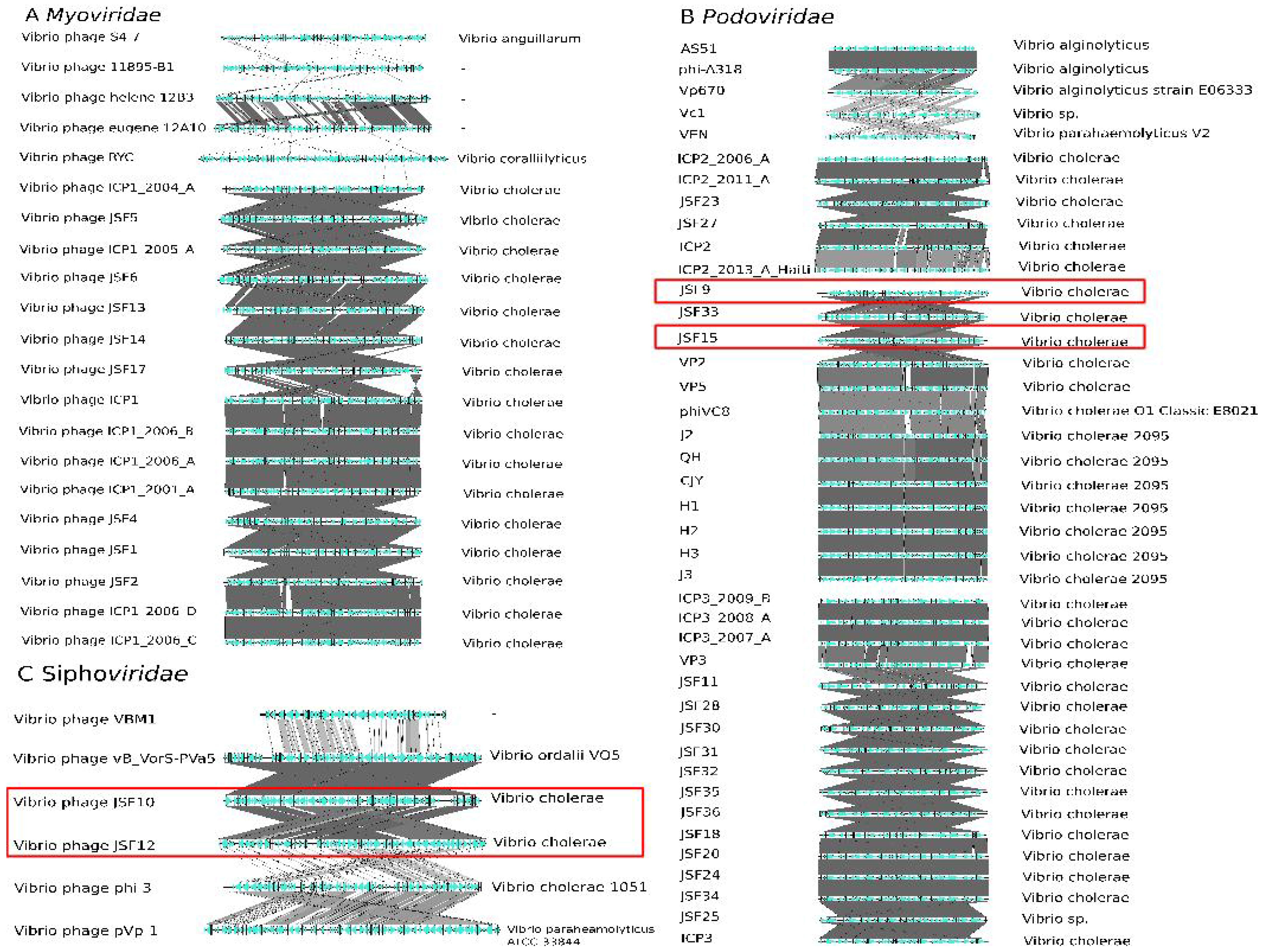
3.5. Inoviridae Taxonomy Phages and Profile HMMs
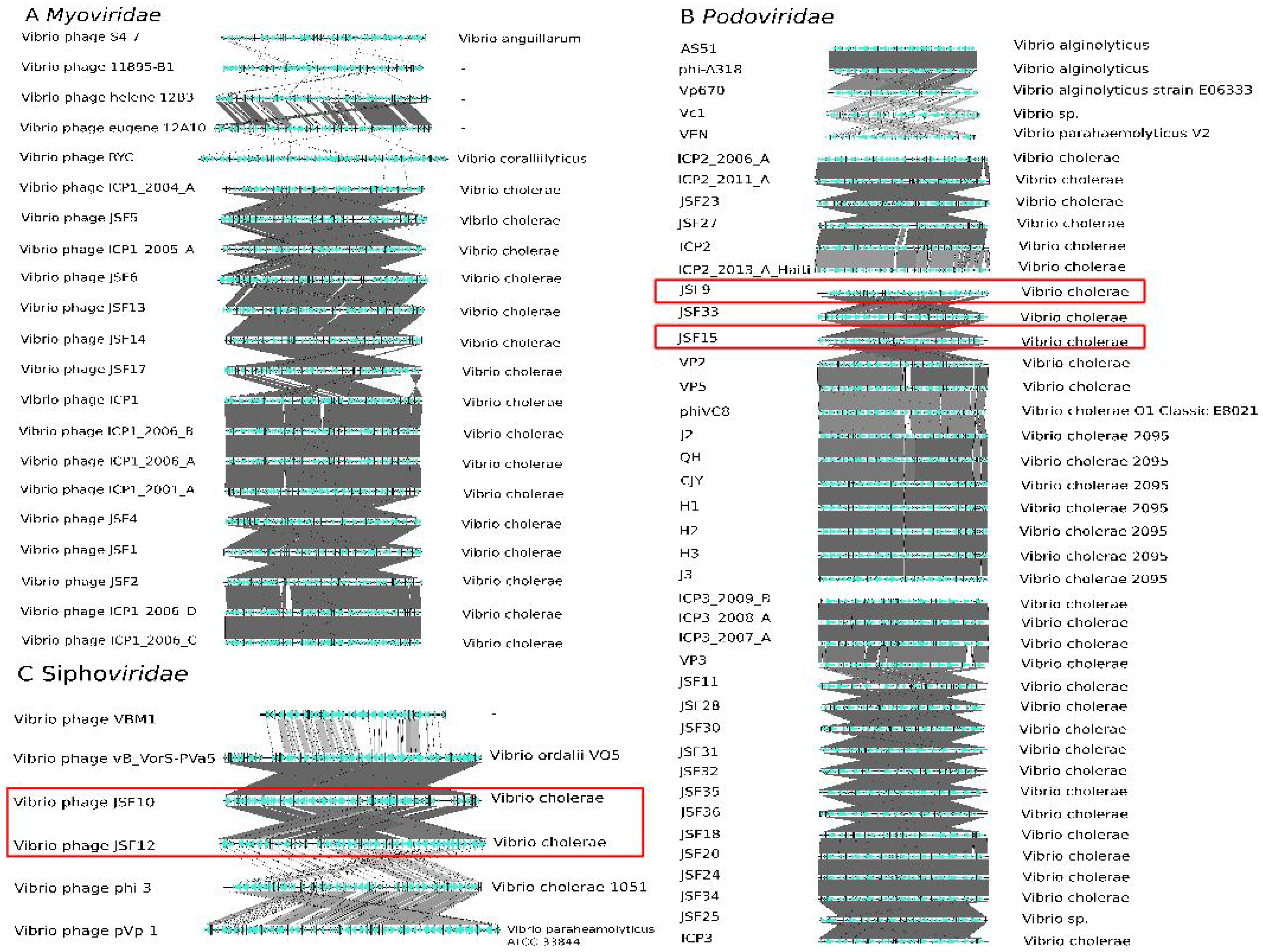
3.6. Taxonomy of Podoviridae
3.7. CDS Prediction and Additional HMM Refinement
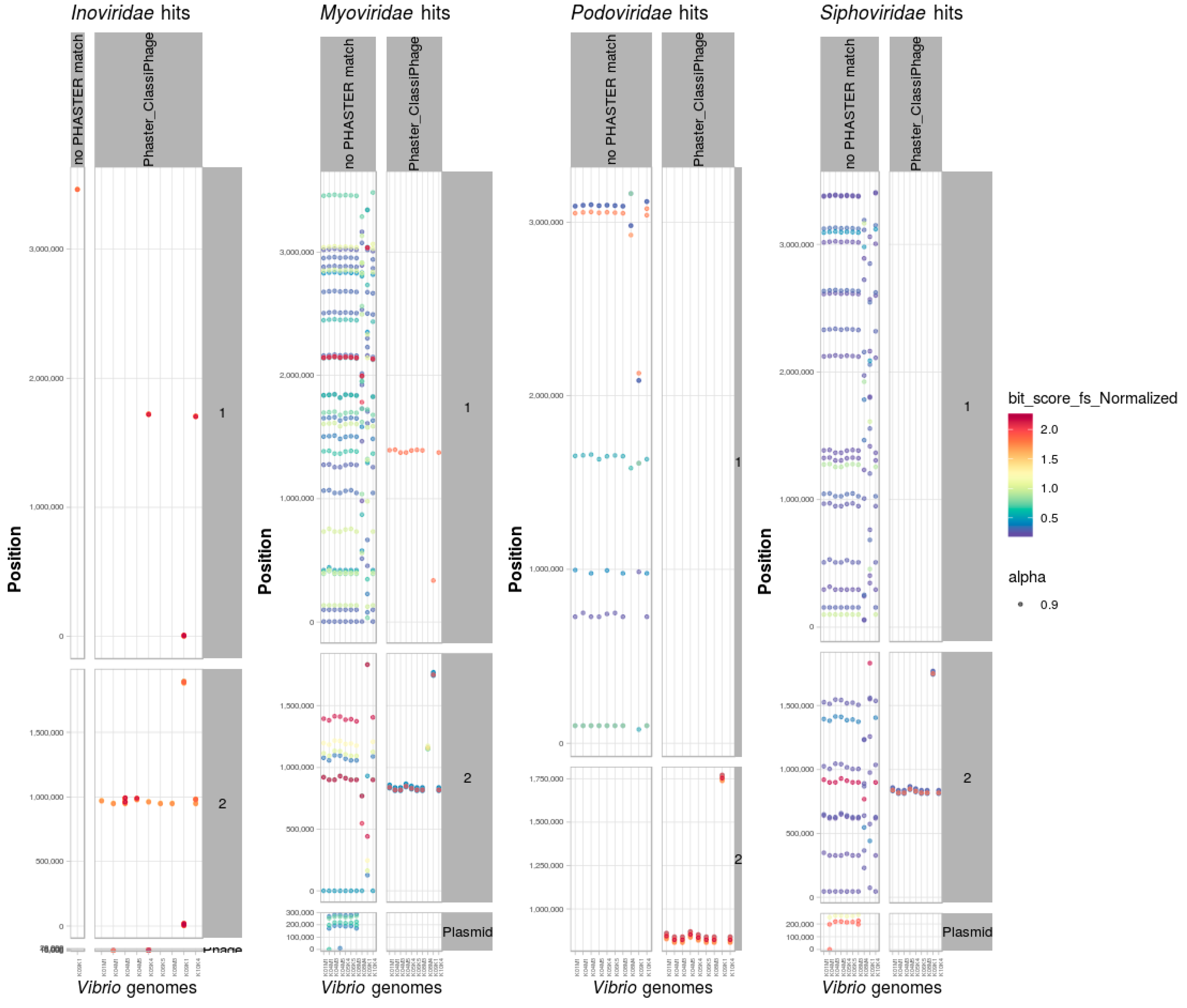
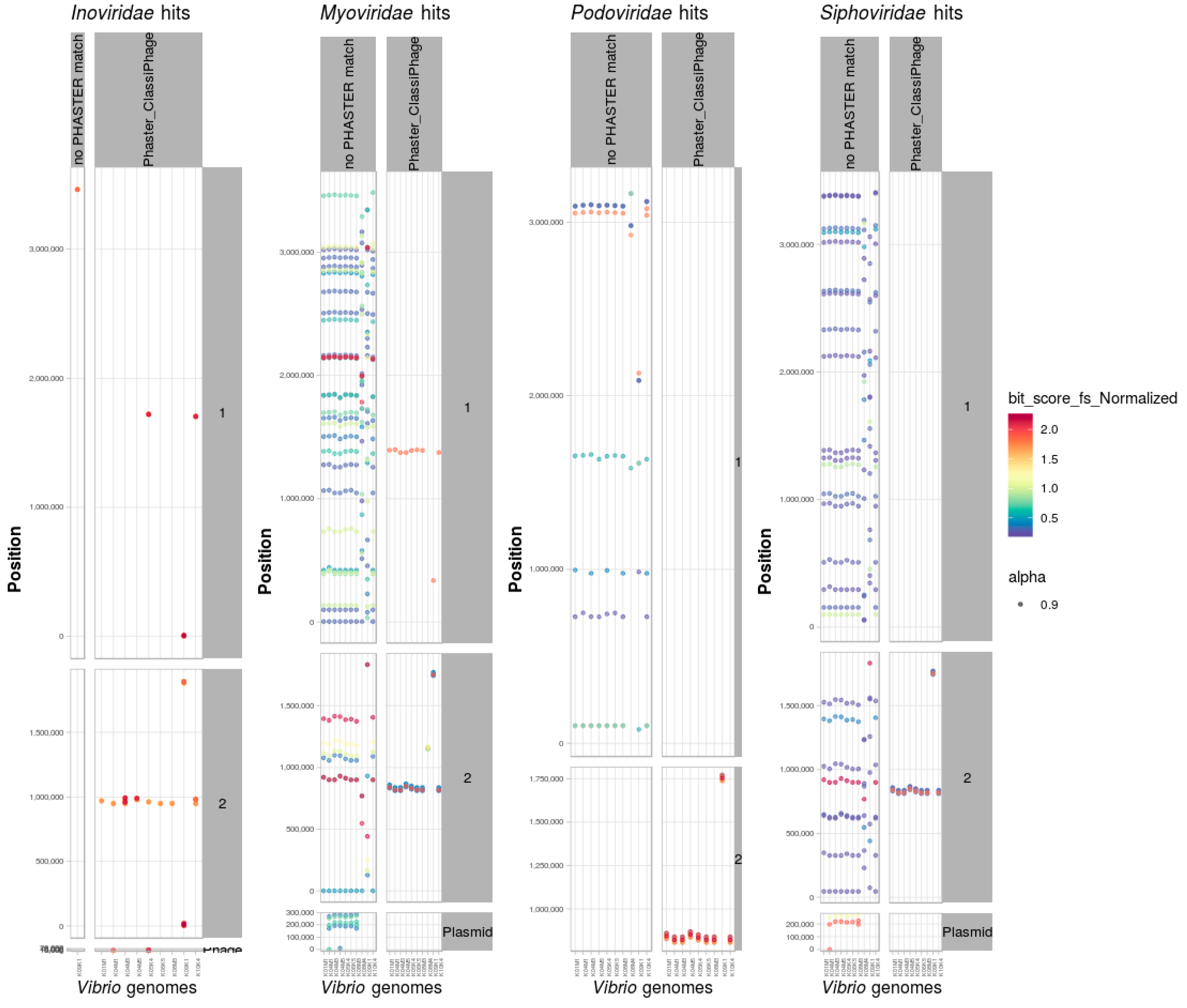
3.8. Identification and Classification of Prophages within Bacterial Genomes
Scan of Positive Dataset of Vibrio Genomes
3.9. PHASTER and ClassiPhage Scan of Published VibrioGenomes, Commonly and Additional Identified Phage Regions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chow, C.-E.T.; Suttle, C.A. Biogeography of Viruses in the Sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses-Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. 50 years of the International Committee on Taxonomy of Viruses: Progress and prospects. Arch. Virol. 2017, 162, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Classification of Bacteriophages. In The Bacteriophages; Calendar, R., Ed.; Oxford University Press: New York, NY, USA, 2006; p. 746. ISBN 9780195148503. [Google Scholar]
- Simmonds, P.; Adams, M.J.; Benk, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Rodney Brister, J. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.W.; Putonti, C. Gene Co-occurrence Networks Reflect Bacteriophage Ecology and Evolution. MBio 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Madslien, E.H.; Olsen, J.S.; Granum, P.E.; Blatny, J.M. Genotyping of B. licheniformis based on a novel multi-locus sequence typing (MLST) scheme. BMC Microbiol. 2012, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Escalona, N.; Jolley, K.A.; Reed, E.; Martinez-Urtaza, J. Defining a Core Genome Multilocus Sequence Typing Scheme for the Global Epidemiology of Vibrio parahaemolyticus. J. Clin. Microbiol. 2017, 55, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Alves, J.M.P.; Durham, A.M.; Gruber, A. Use of profile hidden Markov models in viral discovery: Current insights. Adv. Genomics Genet. 2017, 7, 29–45. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. Elife 2015, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, J.; Krupovic, M.; Koonin, E.V. The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. MBio 2016, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Deschavanne, P.; DuBow, M.S.; Regeard, C. The use of genomic signature distance between bacteriophages and their hosts displays evolutionary relationships and phage growth cycle determination. Virol. J. 2010, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.; Kau, K.; Hussain, F.; Kalatzis, P.; Rørbo, N.; Polz, M.F.; Middelboe, M. Widespread distribution of prophage-encoded virulence factors in marine Vibrio communities. Sci. Rep. 2018, 8, 9973. [Google Scholar] [CrossRef] [PubMed]
- Naser, I.B.; Hoque, M.M.; Abdullah, A.; Bari, S.M.N.; Ghosh, A.N.; Faruque, S.M. Environmental bacteriophages active on biofilms and planktonic forms of toxigenic Vibrio cholerae: Potential relevance in cholera epidemiology. PLoS ONE 2017, 12, e0180838. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J. Gen. Virol. 2018, 99, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Yu, H.J.; Lee, J.H.; Kim, J.-O.; Han, S.H.; Yun, C.-H.; Chun, J.; Nair, G.B.; Kim, D.W. Replication of Vibrio cholerae classical CTX phage. Proc. Natl. Acad. Sci. USA 2017, 114, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Kan, B. Survival and proliferation of the lysogenic bacteriophage CTXΦ in Vibrio cholerae. Virol. Sin. 2015, 30, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Smeal, S.W.; Schmitt, M.A.; Pereira, R.R.; Prasad, A.; Fisk, J.D. Simulation of the M13 life cycle I: Assembly of a genetically-structured deterministic chemical kinetic simulation. Virology 2017, 500, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Mai-Prochnow, A.; Hui, J.G.K.; Kjelleberg, S.; Rakonjac, J.; McDougald, D.; Rice, S.A. Big things in small packages: The genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol. Rev. 2015, 39, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Wendling, C.C.; Piecyk, A.; Refardt, D.; Chibani, C.; Hertel, R.; Liesegang, H.; Bunk, B.; Overmann, J.; Roth, O. Tripartite species interaction: Eukaryotic hosts suffer more from phage susceptible than from phage resistant bacteria. BMC Evol. Biol. 2017, 17, 98. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.J.; Harris, J.B.; Morris, J.G., Jr.; Calderwood, S.B.; Camilli, A. Cholera transmission: The host, pathogen and bacteriophage dynamic. Nat. Rev. Microbiol. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Smeal, S.W.; Schmitt, M.A.; Pereira, R.R.; Prasad, A.; Fisk, J.D. Simulation of the M13 life cycle II: Investigation of the control mechanisms of M13 infection and establishment of the carrier state. Virology 2017, 500, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Senčilo, A.; Luhtanen, A.-M.; Saarijärvi, M.; Bamford, D.H.; Roine, E. Cold-active bacteriophages from the Baltic Sea ice have diverse genomes and virus-host interactions. Environ. Microbiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Doss, J.; Culbertson, K.; Hahn, D.; Camacho, J.; Barekzi, N. A review of phage therapy against bacterial pathogens of aquatic and terrestrial organisms. Viruses 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Gram, L.; Middelboe, M. Vibriophages and their interactions with the fish pathogen Vibrio anguillarum. Appl. Environ. Microbiol. 2014, 80, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.M.P.; De Oliveira, A.L.; Sandberg, T.O.M.; Moreno-Gallego, J.L.; De Toledo, M.A.F.; De Moura, E.M.M.; Oliveira, L.S.; Durham, A.M.; Mehnert, D.U.; De Zanotto, P.M.A.; et al. GenSeed-HMM: A tool for progressive assembly using profile HMMS as seeds and its application in Alpavirinae viral discovery from metagenomic data. Front. Microbiol. 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Day, L.A. Inoviridae. Virus Taxon. 2012, 375–383. [Google Scholar]
- Veesler, D.; Cambillau, C. A Common Evolutionary Origin for Tailed-Bacteriophage Functional Modules and Bacterial Machineries. Microbiol. Mol. Biol. Rev. 2011, 75, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Shin, H.; Ryu, S. Characterization and comparative genomic analysis of bacteriophages infecting members of the Bacillus cereus group. Arch. Virol. 2014, 159, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.S.H.; Waise, T.M.Z.; Kamruzzaman, M.; Ghosh, A.N.; Nair, G.B.; Mekalanos, J.J.; Fanique, S.M. The cyclic AMP (cAMP)-cAMP receptor protein signaling system mediates resistance of Vibrio cholerae O1 strains to multiple environmental bacteriophages. Appl. Environ. Microbiol. 2010, 76, 4233–4240. [Google Scholar] [CrossRef] [PubMed]
- Faruque, S.M.; Mekalanos, J.J. Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 2012, 3, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, G.; Ramaswamy, R. Ab initio gene identification: Prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 2002, 27, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, C.-T. Ori-Finder: A web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinform. 2008, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Linke, B.; McHardy, A.C.; Neuweger, H.; Krause, L.; Meyer, F. REGANOR: A gene prediction server for prokaryotic genomes and a database of high quality gene predictions for prokaryotes. Appl. Bioinform. 2006, 5, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Hertel, R.; Rodríguez, D.P.; Hollensteiner, J.; Dietrich, S.; Leimbach, A.; Hoppert, M.; Liesegang, H.; Volland, S. Genome-Based Identification of Active Prophage Regions by Next Generation Sequencing in Bacillus licheniformis DSM13. PLoS ONE 2015, 10, e0120759. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No of Genomes (Size in Kbp) | No of Proteins | Proteins after MCL | HMMS with >5 Proteins | Positive Evaluated HMMs | |
|---|---|---|---|---|---|
| Siphoviridae | 14 (37.3–128.6) | 1497 | 414 | 94 | 54 |
| Podoviridae | 42 (38.4–112.1) | 2641 | 490 | 233 | 96 |
| Myoviridae | 35 (33.1–250) | 5915 | 921 | 634 | 242 |
| Inoviridae | 19 (6.3–21) | 241 | 39 | 12 | 9 |
| Total | 110 | 10,294 | 1864 | 973 | 401 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chibani, C.M.; Farr, A.; Klama, S.; Dietrich, S.; Liesegang, H. Classifying the Unclassified: A Phage Classification Method. Viruses 2019, 11, 195. https://doi.org/10.3390/v11020195
Chibani CM, Farr A, Klama S, Dietrich S, Liesegang H. Classifying the Unclassified: A Phage Classification Method. Viruses. 2019; 11(2):195. https://doi.org/10.3390/v11020195
Chicago/Turabian StyleChibani, Cynthia Maria, Anton Farr, Sandra Klama, Sascha Dietrich, and Heiko Liesegang. 2019. "Classifying the Unclassified: A Phage Classification Method" Viruses 11, no. 2: 195. https://doi.org/10.3390/v11020195
APA StyleChibani, C. M., Farr, A., Klama, S., Dietrich, S., & Liesegang, H. (2019). Classifying the Unclassified: A Phage Classification Method. Viruses, 11(2), 195. https://doi.org/10.3390/v11020195