Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development

Abstract
Dedication
Abstract
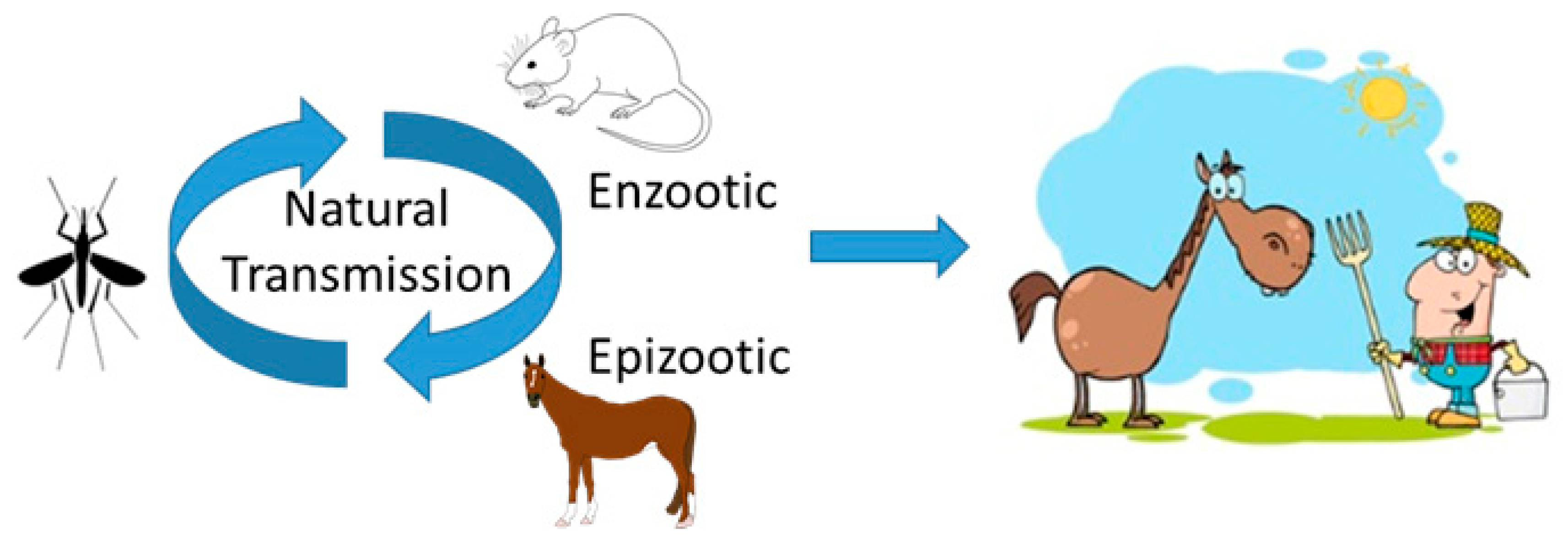
1. Introduction
2. Innate Immune Response to VEEV Infection and the Role of Interferon
3. Role of Alphavirus Genes in Infection
4. Role of Cellular Factors in VEEV Replication
5. CNS Infection of VEEV and the BBB
6. Inflammation in VEEV Infection
7. Vaccines
7.1. Live-Attenuated Vaccine Candidates
7.2. Inactivated Vaccine Candidates
7.3. Chimeric Vaccine Candidates
7.4. Subunit Vaccine Candidate
7.5. Replicon Particles as Vaccine Candidates
7.6. Passive Immunization
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Aguilar, P.V.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Ferro, C.; Haddow, A.D.; Weaver, S.C. Endemic venezuelan equine encephalitis in the americas: Hidden under the dengue umbrella. Future Virol. 2011, 6, 721–740. [Google Scholar] [CrossRef]
- Garoff, H.; Hewson, R.; Opstelten, D.J. Virus maturation by budding. Microbiol. Mol. Biol. Rev. 1998, 62, 1171–1190. [Google Scholar]
- Alan, B.; Weaver, S.C. Arboviruses: Alphaviruses, flaviviruses and bunyaviruses: Encephalitis; yellow fever; dengue; haemorrhagic fever; miscellaneous tropical fevers; undifferentiated fever. In Medical Microbiology, 18th ed.; Elsevier Inc.: London, UK, 2012. [Google Scholar]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.C. Venezuelan equine encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef]
- Eastern equine encephalitis (EEE) virus, venezuelan equine encephalitis (VEE) virus, and western equine encephalitis (WEE) virus. In Biosafety in Microbiological and Biomedical Laboratories; Chosewood, L.C., Wilson, D.E., Eds.; U.S. Department of Health and Human Services/Centers for Disease Control and Prevention/National Institutes of Health, 2009; pp. 242–244. Available online: https://www.cdc.gov/labs/pdf/CDC-BiosafetyMicrobiologicalBiomedicalLaboratories-2009-P.PDF (accessed on 17 February 2019).
- Watts, D.M.; Callahan, J.; Rossi, C.; Oberste, M.S.; Roehrig, J.T.; Wooster, M.T.; Smith, J.F.; Cropp, C.B.; Gentrau, E.M.; Karabatsos, N.; et al. Venezuelan equine encephalitis febrile cases among humans in the peruvian amazon river region. Am. J. Trop. Med. Hyg. 1998, 58, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Rivas, F.; Diaz, L.A.; Cardenas, V.M.; Daza, E.; Bruzon, L.; Alcala, A.; De la Hoz, O.; Caceres, F.M.; Aristizabal, G.; Martinez, J.W.; et al. Epidemic venezuelan equine encephalitis in la guajira, colombia, 1995. J. Infect. Dis. 1997, 175, 828–832. [Google Scholar] [CrossRef]
- Vilcarromero, S.; Aguilar, P.V.; Halsey, E.S.; Laguna-Torres, V.A.; Razuri, H.; Perez, J.; Valderrama, Y.; Gotuzzo, E.; Suarez, L.; Cespedes, M.; et al. Venezuelan equine encephalitis and 2 human deaths, Peru. Emerg. Infect. Dis. 2010, 16, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Ehrenkranz, N.J.; Ventura, A.K. Venezuelan equine encephalitis virus infection in man. Annu. Rev. Med. 1974, 25, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Zacks, M.A.; Paessler, S. Encephalitic alphaviruses. Vet. Microbiol. 2010, 140, 281–286. [Google Scholar] [CrossRef]
- Jackson, A.C.; SenGupta, S.K.; Smith, J.F. Pathogenesis of venezuelan equine encephalitis virus infection in mice and hamsters. Vet. Pathol. 1991, 28, 410–418. [Google Scholar] [CrossRef]
- Charles, P.C.; Walters, E.; Margolis, F.; Johnston, R.E. Mechanism of neuroinvasion of venezuelan equine encephalitis virus in the mouse. Virology 1995, 208, 662–671. [Google Scholar] [CrossRef]
- Vogel, P.; Abplanalp, D.; Kell, W.; Ibrahim, M.S.; Downs, M.B.; Pratt, W.D.; Davis, K.J. Venezuelan equine encephalitis in balb/c mice: Kinetic analysis of central nervous system infection following aerosol or subcutaneous inoculation. Arch. Pathol. Lab. Med. 1996, 120, 164–172. [Google Scholar]
- Schoneboom, B.A.; Catlin, K.M.; Marty, A.M.; Grieder, F.B. Inflammation is a component of neurodegeneration in response to venezuelan equine encephalitis virus infection in mice. J. Neuroimmunol. 2000, 109, 132–146. [Google Scholar] [CrossRef]
- Grieder, F.B.; Davis, N.L.; Aronson, J.F.; Charles, P.C.; Sellon, D.C.; Suzuki, K.; Johnston, R.E. Specific restrictions in the progression of venezuelan equine encephalitis virus-induced disease resulting from single amino acid changes in the glycoproteins. Virology 1995, 206, 994–1006. [Google Scholar] [CrossRef]
- Gupta, P.; Sharma, A.; Han, J.; Yang, A.; Bhomia, M.; Knollmann-Ritschel, B.; Puri, R.K.; Maheshwari, R.K. Differential host gene responses from infection with neurovirulent and partially-neurovirulent strains of venezuelan equine encephalitis virus. BMC Infect. Dis. 2017, 17, 309. [Google Scholar] [CrossRef]
- De la Monte, S.; Castro, F.; Bonilla, N.J.; Gaskin de Urdaneta, A.; Hutchins, G.M. The systemic pathology of venezuelan equine encephalitis virus infection in humans. Am. J. Trop. Med. Hyg. 1985, 34, 194–202. [Google Scholar] [CrossRef]
- Davis, N.L.; Powell, N.; Greenwald, G.F.; Willis, L.V.; Johnson, B.J.; Smith, J.F.; Johnston, R.E. Attenuating mutations in the E2 glycoprotein gene of venezuelan equine encephalitis virus: Construction of single and multiple mutants in a full-length cdna clone. Virology 1991, 183, 20–31. [Google Scholar] [CrossRef]
- Davis, N.L.; Willis, L.V.; Smith, J.F.; Johnston, R.E. In vitro synthesis of infectious venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- Aronson, J.F.; Grieder, F.B.; Davis, N.L.; Charles, P.C.; Knott, T.; Brown, K.; Johnston, R.E. A single-site mutant and revertants arising in vivo define early steps in the pathogenesis of venezuelan equine encephalitis virus. Virology 2000, 270, 111–123. [Google Scholar] [CrossRef]
- Kinney, R.M.; Johnson, B.J.; Welch, J.B.; Tsuchiya, K.R.; Trent, D.W. The full-length nucleotide sequences of the virulent trinidad donkey strain of venezuelan equine encephalitis virus and its attenuated vaccine derivative, strain TC-83. Virology 1989, 170, 19–30. [Google Scholar] [CrossRef]
- Johnston, R.E.; Smith, J.F. Selection for accelerated penetration in cell culture coselects for attenuated mutants of venezuelan equine encephalitis virus. Virology 1988, 162, 437–443. [Google Scholar] [CrossRef]
- Davis, N.L.; Brown, K.W.; Greenwald, G.F.; Zajac, A.J.; Zacny, V.L.; Smith, J.F.; Johnston, R.E. Attenuated mutants of venezuelan equine encephalitis virus containing lethal mutations in the PE2 cleavage signal combined with a second-site suppressor mutation in E1. Virology 1995, 212, 102–110. [Google Scholar] [CrossRef]
- Brandstadter, J.D.; Yang, Y. Natural killer cell responses to viral infection. J. Innate Immun. 2011, 3, 274–279. [Google Scholar] [CrossRef]
- Huprikar, J.; Dal Canto, M.C.; Rabinowitz, S.G. Protection against lethal venezuelan equine encephalitis (VEE) virus infection by cell-free supernatant obtained from immune spleen cells. J. Neurol. Sci. 1990, 97, 143–153. [Google Scholar] [CrossRef]
- Pinto, A.J.; Morahan, P.S.; Brinton, M.A. Comparative study of various immunomodulators for macrophage and natural killer cell activation and antiviral efficacy against exotic RNA viruses. Int. J. Immunopharmacol. 1988, 10, 197–209. [Google Scholar] [CrossRef]
- Saikh, K.U.; Lee, J.S.; Kissner, T.L.; Dyas, B.; Ulrich, R.G. Toll-like receptor and cytokine expression patterns of cd56+ t cells are similar to natural killer cells in response to infection with venezuelan equine encephalitis virus replicons. J. Infect. Dis. 2003, 188, 1562–1570. [Google Scholar] [CrossRef]
- Taylor, K.; Kolokoltsova, O.; Patterson, M.; Poussard, A.; Smith, J.; Estes, D.M.; Paessler, S. Natural killer cell mediated pathogenesis determines outcome of central nervous system infection with venezuelan equine encephalitis virus in C3H/HeN mice. Vaccine 2012, 30, 4095–4105. [Google Scholar] [CrossRef]
- Julander, J.G.; Skirpstunas, R.; Siddharthan, V.; Shafer, K.; Hoopes, J.D.; Smee, D.F.; Morrey, J.D. C3H/HeN mouse model for the evaluation of antiviral agents for the treatment of venezuelan equine encephalitis virus infection. Antiviral Res. 2008, 78, 230–241. [Google Scholar] [CrossRef]
- Steele, K.E.; Davis, K.J.; Stephan, K.; Kell, W.; Vogel, P.; Hart, M.K. Comparative neurovirulence and tissue tropism of wild-type and attenuated strains of venezuelan equine encephalitis virus administered by aerosol in c3h/hen and balb/c mice. Vet. Pathol. 1998, 35, 386–397. [Google Scholar] [CrossRef]
- Gregoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol. Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef]
- Hart, M.K.; Pratt, W.; Panelo, F.; Tammariello, R.; Dertzbaugh, M. Venezuelan equine encephalitis virus vaccines induce mucosal iga responses and protection from airborne infection in BALB/c, but not C3H/HeN mice. Vaccine 1997, 15, 363–369. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Stoermer, K.A.; Morrison, T.E. Complement and viral pathogenesis. Virology 2011, 411, 362–373. [Google Scholar] [CrossRef]
- Brooke, C.B.; Schafer, A.; Matsushima, G.K.; White, L.J.; Johnston, R.E. Early activation of the host complement system is required to restrict central nervous system invasion and limit neuropathology during venezuelan equine encephalitis virus infection. J. Gen. Virol. 2012, 93, 797–806. [Google Scholar] [CrossRef]
- Sharma, A.; Maheshwari, R.K. Oligonucleotide array analysis of toll-like receptors and associated signalling genes in venezuelan equine encephalitis virus-infected mouse brain. J. Gen. Virol. 2009, 90, 1836–1847. [Google Scholar] [CrossRef]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef]
- Valerol, N.; Bonilla, E.; Espina, L.M.; Maldonado, M.; Montero, E.; Anez, F.; Levy, A.; Bermudez, J.; Melean, E.; Nery, A. Increase of interleukin-1 beta, gamma interferon and tumor necrosis factor alpha in serum and brain of mice infected with the venezuelan equine encephalitis virus. Investig. Clin. 2008, 49, 457–467. [Google Scholar]
- Sharma, A.; Bhattacharya, B.; Puri, R.K.; Maheshwari, R.K. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genom. 2008, 9, 289. [Google Scholar] [CrossRef]
- Koterski, J.; Twenhafel, N.; Porter, A.; Reed, D.S.; Martino-Catt, S.; Sobral, B.; Crasta, O.; Downey, T.; DaSilva, L. Gene expression profiling of nonhuman primates exposed to aerosolized venezuelan equine encephalitis virus. FEMS Immunol. Med. Microbiol. 2007, 51, 462–472. [Google Scholar] [CrossRef]
- White, L.J.; Wang, J.G.; Davis, N.L.; Johnston, R.E. Role of alpha/beta interferon in venezuelan equine encephalitis virus pathogenesis: Effect of an attenuating mutation in the 5’ untranslated region. J. Virol. 2001, 75, 3706–3718. [Google Scholar] [CrossRef]
- Grieder, F.B.; Vogel, S.N. Role of interferon and interferon regulatory factors in early protection against venezuelan equine encephalitis virus infection. Virology 1999, 257, 106–118. [Google Scholar] [CrossRef]
- Simmons, J.D.; White, L.J.; Morrison, T.E.; Montgomery, S.A.; Whitmore, A.C.; Johnston, R.E.; Heise, M.T. Venezuelan equine encephalitis virus disrupts stat1 signaling by distinct mechanisms independent of host shutoff. J. Virol. 2009, 83, 10571–10581. [Google Scholar] [CrossRef]
- Yin, J.; Gardner, C.L.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Similarities and differences in antagonism of neuron alpha/beta interferon responses by venezuelan equine encephalitis and sindbis alphaviruses. J. Virol. 2009, 83, 10036–10047. [Google Scholar] [CrossRef]
- Hefti, E.; Bishop, D.H.; Dubin, D.T.; Stollar, V. 5’ nucleotide sequence of sindbis viral RNA. J. Virol. 1975, 17, 149–159. [Google Scholar]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar]
- Frolov, I.; Akhrymuk, M.; Akhrymuk, I.; Atasheva, S.; Frolova, E.I. Early events in alphavirus replication determine the outcome of infection. J. Virol. 2012, 86, 5055–5066. [Google Scholar] [CrossRef]
- Garmashova, N.; Atasheva, S.; Kang, W.; Weaver, S.C.; Frolova, E.; Frolov, I. Analysis of venezuelan equine encephalitis virus capsid protein function in the inhibition of cellular transcription. J. Virol. 2007, 81, 13552–13565. [Google Scholar] [CrossRef]
- Garmashova, N.; Gorchakov, R.; Frolova, E.; Frolov, I. Sindbis virus nonstructural protein NSP2 is cytotoxic and inhibits cellular transcription. J. Virol. 2006, 80, 5686–5696. [Google Scholar] [CrossRef]
- Garmashova, N.; Gorchakov, R.; Volkova, E.; Paessler, S.; Frolova, E.; Frolov, I. The old world and new world alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 2007, 81, 2472–2484. [Google Scholar] [CrossRef]
- Atasheva, S.; Fish, A.; Fornerod, M.; Frolova, E.I. Venezuelan equine encephalitis virus capsid protein forms a tetrameric complex with crm1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol. 2010, 84, 4158–4171. [Google Scholar] [CrossRef]
- Atasheva, S.; Garmashova, N.; Frolov, I.; Frolova, E. Venezuelan equine encephalitis virus capsid protein inhibits nuclear import in mammalian but not in mosquito cells. J. Virol. 2008, 82, 4028–4041. [Google Scholar] [CrossRef]
- Lundberg, L.; Pinkham, C.; Baer, A.; Amaya, M.; Narayanan, A.; Wagstaff, K.M.; Jans, D.A.; Kehn-Hall, K. Nuclear import and export inhibitors alter capsid protein distribution in mammalian cells and reduce venezuelan equine encephalitis virus replication. Antiviral Res. 2013, 100, 662–672. [Google Scholar] [CrossRef]
- Atasheva, S.; Krendelchtchikova, V.; Liopo, A.; Frolova, E.; Frolov, I. Interplay of acute and persistent infections caused by venezuelan equine encephalitis virus encoding mutated capsid protein. J. Virol. 2010, 84, 10004–10015. [Google Scholar] [CrossRef]
- Bhalla, N.; Sun, C.; Metthew Lam, L.K.; Gardner, C.L.; Ryman, K.D.; Klimstra, W.B. Host translation shutoff mediated by non-structural protein 2 is a critical factor in the antiviral state resistance of venezuelan equine encephalitis virus. Virology 2016, 496, 147–165. [Google Scholar] [CrossRef]
- Snyder, J.E.; Kulcsar, K.A.; Schultz, K.L.; Riley, C.P.; Neary, J.T.; Marr, S.; Jose, J.; Griffin, D.E.; Kuhn, R.J. Functional characterization of the alphavirus TF protein. J. Virol. 2013, 87, 8511–8523. [Google Scholar] [CrossRef]
- Kendra, J.A.; de la Fuente, C.; Brahms, A.; Woodson, C.; Bell, T.M.; Chen, B.; Khan, Y.A.; Jacobs, J.L.; Kehn-Hall, K.; Dinman, J.D. Ablation of programmed −1 ribosomal frameshifting in venezuelan equine encephalitis virus results in attenuated neuropathogenicity. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Miller, D.J. Role of cellular lipids in positive-sense rna virus replication complex assembly and function. Viruses 2010, 2, 1055–1068. [Google Scholar] [CrossRef]
- Volkova, E.; Gorchakov, R.; Frolov, I. The efficient packaging of venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology 2006, 344, 315–327. [Google Scholar] [CrossRef]
- Michel, G.; Petrakova, O.; Atasheva, S.; Frolov, I. Adaptation of venezuelan equine encephalitis virus lacking 51-nt conserved sequence element to replication in mammalian and mosquito cells. Virology 2007, 362, 475–487. [Google Scholar] [CrossRef]
- Petrakova, O.; Volkova, E.; Gorchakov, R.; Paessler, S.; Kinney, R.M.; Frolov, I. Noncytopathic replication of venezuelan equine encephalitis virus and eastern equine encephalitis virus replicons in mammalian cells. J. Virol. 2005, 79, 7597–7608. [Google Scholar] [CrossRef]
- Li, C.; Guillen, J.; Rabah, N.; Blanjoie, A.; Debart, F.; Vasseur, J.J.; Canard, B.; Decroly, E.; Coutard, B. Mrna capping by venezuelan equine encephalitis virus nsp1: Functional characterization and implications for antiviral research. J. Virol. 2015, 89, 8292–8303. [Google Scholar] [CrossRef]
- Hardy, W.R.; Strauss, J.H. Processing the nonstructural polyproteins of sindbis virus: Nonstructural proteinase is in the c-terminal half of NSP2 and functions both in cis and in trans. J. Virol. 1989, 63, 4653–4664. [Google Scholar]
- Merits, A.; Vasiljeva, L.; Ahola, T.; Kaariainen, L.; Auvinen, P. Proteolytic processing of semliki forest virus-specific non-structural polyprotein by NSP2 protease. J. Gen. Virol. 2001, 82, 765–773. [Google Scholar] [CrossRef]
- Mayuri; Geders, T.W.; Smith, J.L.; Kuhn, R.J. Role for conserved residues of sindbis virus nonstructural protein 2 methyltransferase-like domain in regulation of minus-strand synthesis and development of cytopathic infection. J. Virol. 2008, 82, 7284–7297. [Google Scholar] [CrossRef]
- Montgomery, S.A.; Johnston, R.E. Nuclear import and export of venezuelan equine encephalitis virus nonstructural protein 2. J. Virol. 2007, 81, 10268–10279. [Google Scholar] [CrossRef]
- Kim, D.Y.; Atasheva, S.; Frolova, E.I.; Frolov, I. Venezuelan equine encephalitis virus NSP2 protein regulates packaging of the viral genome into infectious virions. J. Virol. 2013, 87, 4202–4213. [Google Scholar] [CrossRef]
- Foy, N.J.; Akhrymuk, M.; Shustov, A.V.; Frolova, E.I.; Frolov, I. Hypervariable domain of nonstructural protein nsp3 of venezuelan equine encephalitis virus determines cell-specific mode of virus replication. J. Virol. 2013, 87, 7569–7584. [Google Scholar] [CrossRef]
- Foy, N.J.; Akhrymuk, M.; Akhrymuk, I.; Atasheva, S.; Bopda-Waffo, A.; Frolov, I.; Frolova, E.I. Hypervariable domains of NSP3 proteins of new world and old world alphaviruses mediate formation of distinct, virus-specific protein complexes. J. Virol. 2013, 87, 1997–2010. [Google Scholar] [CrossRef]
- Rupp, J.C.; Jundt, N.; Hardy, R.W. Requirement for the amino-terminal domain of sindbis virus NSP4 during virus infection. J. Virol. 2011, 85, 3449–3460. [Google Scholar] [CrossRef]
- Fata, C.L.; Sawicki, S.G.; Sawicki, D.L. Alphavirus minus-strand rna synthesis: Identification of a role for Arg183 of the NSP4 polymerase. J. Virol. 2002, 76, 8632–8640. [Google Scholar] [CrossRef]
- Takkinen, K.; Peranen, J.; Keranen, S.; Soderlund, H.; Kaariainen, L. The semliki-forest-virus-specific nonstructural protein NSP4 is an autoproteinase. Eur. J. Biochem. 1990, 189, 33–38. [Google Scholar] [CrossRef]
- Tomar, S.; Hardy, R.W.; Smith, J.L.; Kuhn, R.J. Catalytic core of alphavirus nonstructural protein NSP4 possesses terminal adenylyltransferase activity. J. Virol. 2006, 80, 9962–9969. [Google Scholar] [CrossRef]
- Malygin, A.A.; Bondarenko, E.I.; Ivanisenko, V.A.; Protopopova, E.V.; Karpova, G.G.; Loktev, V.B. C-terminal fragment of human laminin-binding protein contains a receptor domain for venezuelan equine encephalitis and tick-borne encephalitis viruses. Biochemistry (Mosc) 2009, 74, 1328–1336. [Google Scholar] [CrossRef]
- Ludwig, G.V.; Kondig, J.P.; Smith, J.F. A putative receptor for venezuelan equine encephalitis virus from mosquito cells. J. Virol. 1996, 70, 5592–5599. [Google Scholar]
- Kolokoltsov, A.A.; Fleming, E.H.; Davey, R.A. Venezuelan equine encephalitis virus entry mechanism requires late endosome formation and resists cell membrane cholesterol depletion. Virology 2006, 347, 333–342. [Google Scholar] [CrossRef]
- Bernard, K.A.; Klimstra, W.B.; Johnston, R.E. Mutations in the E2 glycoprotein of venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 2000, 276, 93–103. [Google Scholar] [CrossRef]
- Gardner, C.L.; Hritz, J.; Sun, C.; Vanlandingham, D.L.; Song, T.Y.; Ghedin, E.; Higgs, S.; Klimstra, W.B.; Ryman, K.D. Deliberate attenuation of chikungunya virus by adaptation to heparan sulfate-dependent infectivity: A model for rational arboviral vaccine design. PLoS Negl. Trop. Dis. 2014, 8, e2719. [Google Scholar] [CrossRef]
- Gardner, C.L.; Choi-Nurvitadhi, J.; Sun, C.; Bayer, A.; Hritz, J.; Ryman, K.D.; Klimstra, W.B. Natural variation in the heparan sulfate binding domain of the eastern equine encephalitis virus e2 glycoprotein alters interactions with cell surfaces and virulence in mice. J. Virol. 2013, 87, 8582–8590. [Google Scholar] [CrossRef]
- Chelladurai, P.; Seeger, W.; Pullamsetti, S.S. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur. Respir. J. 2012, 40, 766–782. [Google Scholar] [CrossRef]
- Ryman, K.D.; Gardner, C.L.; Burke, C.W.; Meier, K.C.; Thompson, J.M.; Klimstra, W.B. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted sindbis viruses. J. Virol. 2007, 81, 3563–3573. [Google Scholar] [CrossRef]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; de la Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The role of ikkbeta in venezuelan equine encephalitis virus infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O’Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef]
- Kim, D.Y.; Reynaud, J.M.; Rasalouskaya, A.; Akhrymuk, I.; Mobley, J.A.; Frolov, I.; Frolova, E.I. New world and old world alphaviruses have evolved to exploit different components of stress granules, FXR and G3BP proteins, for assembly of viral replication complexes. PLoS Pathog. 2016, 12, e1005810. [Google Scholar] [CrossRef]
- Amaya, M.; Brooks-Faulconer, T.; Lark, T.; Keck, F.; Bailey, C.; Raman, V.; Narayanan, A. Venezuelan equine encephalitis virus non-structural protein 3 (nsp3) interacts with RNA helicases DDX1 and DDX3 in infected cells. Antiviral Res. 2016, 131, 49–60. [Google Scholar] [CrossRef]
- Atasheva, S.; Akhrymuk, M.; Frolova, E.I.; Frolov, I. New parp gene with an anti-alphavirus function. J. Virol. 2012, 86, 8147–8160. [Google Scholar] [CrossRef]
- Atasheva, S.; Frolova, E.I.; Frolov, I. Interferon-stimulated poly(adp-ribose) polymerases are potent inhibitors of cellular translation and virus replication. J. Virol. 2014, 88, 2116–2130. [Google Scholar] [CrossRef]
- Poddar, S.; Hyde, J.L.; Gorman, M.J.; Farzan, M.; Diamond, M.S. The interferon-stimulated gene IFITM3 restricts infection and pathogenesis of arthritogenic and encephalitic alphaviruses. J. Virol. 2016, 90, 8780–8794. [Google Scholar] [CrossRef]
- Muehlenbein, M.P.; Cogswell, F.B.; James, M.A.; Koterski, J.; Ludwig, G.V. Testosterone correlates with venezuelan equine encephalitis virus infection in macaques. Virol. J. 2006, 3, 19. [Google Scholar] [CrossRef]
- Ryzhikov, A.B.; Ryabchikova, E.I.; Sergeev, A.N.; Tkacheva, N.V. Spread of venezuelan equine encephalitis virus in mice olfactory tract. Arch. Virol. 1995, 140, 2243–2254. [Google Scholar] [CrossRef]
- Park, C.H.; Ishinaka, M.; Takada, A.; Kida, H.; Kimura, T.; Ochiai, K.; Umemura, T. The invasion routes of neurovirulent a/hong kong/483/97 (H5N1) influenza virus into the central nervous system after respiratory infection in mice. Arch. Virol. 2002, 147, 1425–1436. [Google Scholar] [CrossRef]
- Harberts, E.; Yao, K.; Wohler, J.E.; Maric, D.; Ohayon, J.; Henkin, R.; Jacobson, S. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 13734–13739. [Google Scholar] [CrossRef]
- Faber, H.K.; Silverberg, R.J.; Dong, L. Poliomyelitis in the cynomolgus monkey: Iii. Infection by inhalation of droplet nuclei and the nasopharyngeal portal of entry, with a note on this mode of infection in rhesus. J. Exp. Med. 1944, 80, 39–57. [Google Scholar] [CrossRef]
- Plakhov, I.V.; Arlund, E.E.; Aoki, C.; Reiss, C.S. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology 1995, 209, 257–262. [Google Scholar] [CrossRef]
- Lafay, F.; Coulon, P.; Astic, L.; Saucier, D.; Riche, D.; Holley, A.; Flamand, A. Spread of the CVS strain of rabies virus and of the avirulent mutant avo1 along the olfactory pathways of the mouse after intranasal inoculation. Virology 1991, 183, 320–330. [Google Scholar] [CrossRef]
- Yamada, M.; Nakamura, K.; Yoshii, M.; Kaku, Y.; Narita, M. Brain lesions induced by experimental intranasal infection of Japanese encephalitis virus in piglets. J. Comp. Pathol. 2009, 141, 156–162. [Google Scholar] [CrossRef]
- Gorelkin, L. Venezuelan equine encephalomyelitis in an adult animal host. An electron microscopic study. Am. J. Pathol. 1973, 73, 425–442. [Google Scholar]
- Schafer, A.; Brooke, C.B.; Whitmore, A.C.; Johnston, R.E. The role of the blood-brain barrier during venezuelan equine encephalitis virus infection. J. Virol. 2011, 85, 10682–10690. [Google Scholar] [CrossRef]
- Steele, K.E.; Twenhafel, N.A. Review paper: Pathology of animal models of alphavirus encephalitis. Vet. Pathol. 2010, 47, 790–805. [Google Scholar] [CrossRef]
- Schafer, A.; Whitmore, A.C.; Konopka, J.L.; Johnston, R.E. Replicon particles of venezuelan equine encephalitis virus as a reductionist murine model for encephalitis. J. Virol. 2009, 83, 4275–4286. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef]
- Steele, K.E.; Seth, P.; Catlin-Lebaron, K.M.; Schoneboom, B.A.; Husain, M.M.; Grieder, F.; Maheshwari, R.K. Tunicamycin enhances neuroinvasion and encephalitis in mice infected with venezuelan equine encephalitis virus. Vet. Pathol. 2006, 43, 904–913. [Google Scholar] [CrossRef]
- Shukla, A.; Shukla, G.S.; Srimal, R.C. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 1996, 15, 400–405. [Google Scholar] [CrossRef]
- Seth, P.; Husain, M.M.; Gupta, P.; Schoneboom, A.; Grieder, B.F.; Mani, H.; Maheshwari, R.K. Early onset of virus infection and up-regulation of cytokines in mice treated with cadmium and manganese. Biometals 2003, 16, 359–368. [Google Scholar] [CrossRef]
- Jackson, A.C.; Rossiter, J.P. Apoptotic cell death is an important cause of neuronal injury in experimental venezuelan equine encephalitis virus infection of mice. Acta Neuropathol. 1997, 93, 349–353. [Google Scholar] [CrossRef]
- Schoneboom, B.A.; Fultz, M.J.; Miller, T.H.; McKinney, L.C.; Grieder, F.B. Astrocytes as targets for venezuelan equine encephalitis virus infection. J. Neurovirol. 1999, 5, 342–354. [Google Scholar] [CrossRef]
- Schoneboom, B.A.; Lee, J.S.; Grieder, F.B. Early expression of IFN-alpha/beta and inos in the brains of venezuelan equine encephalitis virus-infected mice. J. Interferon Cytokine Res. 2000, 20, 205–215. [Google Scholar] [CrossRef]
- Keck, F.; Brooks-Faulconer, T.; Lark, T.; Ravishankar, P.; Bailey, C.; Salvador-Morales, C.; Narayanan, A. Altered mitochondrial dynamics as a consequence of venezuelan equine encephalitis virus infection. Virulence 2017, 8, 1849–1866. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. ICAM-1: Role in inflammation and in the regulation of vascular permeability. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H926–H927. [Google Scholar] [CrossRef]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; Lopez-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 2002, 100, 1160–1167. [Google Scholar]
- Zhang, K.; McQuibban, G.A.; Silva, C.; Butler, G.S.; Johnston, J.B.; Holden, J.; Clark-Lewis, I.; Overall, C.M.; Power, C. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 2003, 6, 1064–1071. [Google Scholar] [CrossRef]
- Mohan, M.J.; Seaton, T.; Mitchell, J.; Howe, A.; Blackburn, K.; Burkhart, W.; Moyer, M.; Patel, I.; Waitt, G.M.; Becherer, J.D.; et al. The tumor necrosis factor-alpha converting enzyme (tace): A unique metalloproteinase with highly defined substrate selectivity. Biochemistry 2002, 41, 9462–9469. [Google Scholar] [CrossRef]
- Schonbeck, U.; Mach, F.; Libby, P. Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar]
- Maeda, S.; Dean, D.D.; Gomez, R.; Schwartz, Z.; Boyan, B.D. The first stage of transforming growth factor beta1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (mmp-3). Calcif. Tissue Int. 2002, 70, 54–65. [Google Scholar] [CrossRef]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and immune modulation of venezuelan equine encephalitis virus-induced disease in the mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef]
- Berge, T.O.; Banks, I.S.; Tigertt, W.D. Attenuation of venezuelan equine encephalomyelitis virus by in vitro cultivation in guinea-pig heart cells. Am. J. Hyg. 1961, 73, 209–218. [Google Scholar]
- Kinney, R.M.; Chang, G.J.; Tsuchiya, K.R.; Sneider, J.M.; Roehrig, J.T.; Woodward, T.M.; Trent, D.W. Attenuation of venezuelan equine encephalitis virus strain tc-83 is encoded by the 5’-noncoding region and the E2 envelope glycoprotein. J. Virol. 1993, 67, 1269–1277. [Google Scholar]
- Pittman, P.R.; Makuch, R.S.; Mangiafico, J.A.; Cannon, T.L.; Gibbs, P.H.; Peters, C.J. Long-term duration of detectable neutralizing antibodies after administration of live-attenuated vee vaccine and following booster vaccination with inactivated vee vaccine. Vaccine 1996, 14, 337–343. [Google Scholar] [CrossRef]
- Paessler, S.; Weaver, S.C. Vaccines for venezuelan equine encephalitis. Vaccine 2009, 27, D80–D85. [Google Scholar] [CrossRef]
- Alevizatos, A.C.; McKinney, R.W.; Feigin, R.D. Live, attenuated venezuelan equine encephalomyelitis virus vaccine. I. Clinical effects in man. Am. J. Trop Med. Hyg. 1967, 16, 762–768. [Google Scholar] [CrossRef]
- Pedersen, C.E., Jr.; Robinson, D.M.; Cole, F.E., Jr. Isolation of the vaccine strain of venezuelan equine encephalomyelitis virus from mosquitoes in louisiana. Am. J. Epidemiol. 1972, 95, 490–496. [Google Scholar] [CrossRef]
- Erwin-Cohen, R.; Porter, A.; Pittman, P.; Rossi, C.; Dasilva, L. Host responses to live-attenuated venezuelan equine encephalitis virus (TC-83): Comparison of naive, vaccine responder and nonresponder to TC-83 challenge in human peripheral blood mononuclear cells. Hum. Vaccin. Immunother. 2012, 8, 1053–1065. [Google Scholar] [CrossRef]
- Pratt, W.D.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Genetically engineered, live attenuated vaccines for venezuelan equine encephalitis: Testing in animal models. Vaccine 2003, 21, 3854–3862. [Google Scholar] [CrossRef]
- Rao, V.; Hinz, M.E.; Roberts, B.A.; Fine, D. Environmental hazard assessment of venezuelan equine encephalitis virus vaccine candidate strain V3526. Vaccine 2004, 22, 2667–2673. [Google Scholar] [CrossRef]
- Fine, D.L.; Roberts, B.A.; Teehee, M.L.; Terpening, S.J.; Kelly, C.L.; Raetz, J.L.; Baker, D.C.; Powers, A.M.; Bowen, R.A. Venezuelan equine encephalitis virus vaccine candidate (V3526) safety, immunogenicity and efficacy in horses. Vaccine 2007, 25, 1868–1876. [Google Scholar] [CrossRef]
- Reed, D.S.; Lind, C.M.; Lackemeyer, M.G.; Sullivan, L.J.; Pratt, W.D.; Parker, M.D. Genetically engineered, live, attenuated vaccines protect nonhuman primates against aerosol challenge with a virulent IE strain of venezuelan equine encephalitis virus. Vaccine 2005, 23, 3139–3147. [Google Scholar] [CrossRef]
- Holley, P.; Fine, D.L.; Terpening, S.J.; Mallory, C.J.; Main, C.A.; Snow, D.M. Safety of an attenuated venezuelan equine encephalitis virus (VEEV) vaccine in humans. In Proceedings of the 48th ICAAC/IDSA, Washington, DC, USA, 25–28 October 2008. [Google Scholar]
- Sharma, A.; Gupta, P.; Glass, P.J.; Parker, M.D.; Maheshwari, R.K. Safety and protective efficacy of ina-inactivated venezuelan equine encephalitis virus: Implication in vaccine development. Vaccine 2011, 29, 953–959. [Google Scholar] [CrossRef][Green Version]
- Cole, F.E., Jr.; May, S.W.; Robinson, D.M. Formalin-inactivated venezuelan equine encephalomyelitis (trinidad strain) vaccine produced in rolling-bottle cultures of chicken embryo cells. Appl. Microbiol. 1973, 25, 262–265. [Google Scholar] [PubMed]
- Kinney, R.M.; Tsuchiya, K.R.; Sneider, J.M.; Trent, D.W. Molecular evidence for the origin of the widespread venezuelan equine encephalitis epizootic of 1969 to 1972. J. Gen. Virol. 1992, 73, 3301–3305. [Google Scholar] [CrossRef]
- Weaver, S.C.; Pfeffer, M.; Marriott, K.; Kang, W.; Kinney, R.M. Genetic evidence for the origins of venezuelan equine encephalitis virus subtype IAB outbreaks. Am. J. Trop Med. Hyg. 1999, 60, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Edelman, R.; Ascher, M.S.; Oster, C.N.; Ramsburg, H.H.; Cole, F.E.; Eddy, G.A. Evaluation in humans of a new, inactivated vaccine for venezuelan equine encephalitis virus (C-84). J. Infect. Dis. 1979, 140, 708–715. [Google Scholar] [CrossRef]
- Martin, S.S.; Bakken, R.R.; Lind, C.M.; Garcia, P.; Jenkins, E.; Glass, P.J.; Parker, M.D.; Hart, M.K.; Fine, D.L. Evaluation of formalin inactivated v3526 virus with adjuvant as a next generation vaccine candidate for venezuelan equine encephalitis virus. Vaccine 2010, 28, 3143–3151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fine, D.L.; Jenkins, E.; Martin, S.S.; Glass, P.; Parker, M.D.; Grimm, B. A multisystem approach for development and evaluation of inactivated vaccines for venezuelan equine encephalitis virus (VEEV). J. Virol. Methods 2010, 163, 424–432. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sharma, A.; Raviv, Y.; Puri, A.; Viard, M.; Blumenthal, R.; Maheshwari, R.K. Complete inactivation of venezuelan equine encephalitis virus by 1,5-iodonaphthylazide. Biochem. Biophys. Res. Commun. 2007, 358, 392–398. [Google Scholar] [CrossRef]
- Gupta, P.; Sharma, A.; Mathias, V.; Raviv, Y.; Blumenthal, R.; Maheshwari, R.K. Inactivation of non-enveloped virus by 1,5 iodonaphthyl azide. BMC Res. Notes 2015, 8, 44. [Google Scholar] [CrossRef][Green Version]
- Gupta, P.; Sharma, A.; Spurgers, K.B.; Bakken, R.R.; Eccleston, L.T.; Cohen, J.W.; Honnold, S.P.; Glass, P.J.; Maheshwari, R.K. 1,5-iodonaphthyl azide-inactivated V3526 protects against aerosol challenge with virulent venezuelan equine encephalitis virus. Vaccine 2016, 34, 2762–2765. [Google Scholar] [CrossRef]
- Martin, S.S.; Bakken, R.R.; Lind, C.M.; Garcia, P.; Jenkins, E.; Glass, P.J.; Parker, M.D.; Hart, M.K.; Fine, D.L. Comparison of the immunological responses and efficacy of gamma-irradiated V3526 vaccine formulations against subcutaneous and aerosol challenge with venezuelan equine encephalitis virus subtype IAB. Vaccine 2010, 28, 1031–1040. [Google Scholar] [CrossRef]
- Gaidamakova, E.K.; Myles, I.A.; McDaniel, D.P.; Fowler, C.J.; Valdez, P.A.; Naik, S.; Gayen, M.; Gupta, P.; Sharma, A.; Glass, P.J.; et al. Preserving immunogenicity of lethally irradiated viral and bacterial vaccine epitopes using a radio- protective Mn2+-peptide complex from deinococcus. Cell Host Microbe 2012, 12, 117–124. [Google Scholar] [CrossRef]
- Gayen, M.; Gupta, P.; Morazzani, E.M.; Gaidamakova, E.K.; Knollmann-Ritschel, B.; Daly, M.J.; Glass, P.J.; Maheshwari, R.K. Deinococcus Mn2+-peptide complex: A novel approach to alphavirus vaccine development. Vaccine 2017, 35, 3672–3681. [Google Scholar] [CrossRef]
- Biologics, C.f.V. Chimera as an additional naming convention for live recombinant products. In CENTER FOR VETERINARY BIOLOGICS NOTICE NO. 05-23; United States Department of Agriculture: Ames, IA, USA, 2005; p. 2. [Google Scholar]
- Volkova, E.; Frolova, E.; Darwin, J.R.; Forrester, N.L.; Weaver, S.C.; Frolov, I. IRES-dependent replication of venezuelan equine encephalitis virus makes it highly attenuated and incapable of replicating in mosquito cells. Virology 2008, 377, 160–169. [Google Scholar] [CrossRef][Green Version]
- Guerbois, M.; Volkova, E.; Forrester, N.L.; Rossi, S.L.; Frolov, I.; Weaver, S.C. IRES-driven expression of the capsid protein of the venezuelan equine encephalitis virus tc-83 vaccine strain increases its attenuation and safety. PLoS Negl. Trop Dis. 2013, 7, e2197. [Google Scholar] [CrossRef]
- Rossi, S.L.; Guerbois, M.; Gorchakov, R.; Plante, K.S.; Forrester, N.L.; Weaver, S.C. Ires-based venezuelan equine encephalitis vaccine candidate elicits protective immunity in mice. Virology 2013, 437, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Kim, D.Y.; Frolova, E.I.; Frolov, I. Venezuelan equine encephalitis virus variants lacking transcription inhibitory functions demonstrate highly attenuated phenotype. J. Virol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Paessler, S.; Fayzulin, R.Z.; Anishchenko, M.; Greene, I.P.; Weaver, S.C.; Frolov, I. Recombinant sindbis/venezuelan equine encephalitis virus is highly attenuated and immunogenic. J. Virol. 2003, 77, 9278–9286. [Google Scholar] [CrossRef]
- Paessler, S.; Ni, H.; Petrakova, O.; Fayzulin, R.Z.; Yun, N.; Anishchenko, M.; Weaver, S.C.; Frolov, I. Replication and clearance of venezuelan equine encephalitis virus from the brains of animals vaccinated with chimeric SIN/VEE viruses. J. Virol. 2006, 80, 2784–2796. [Google Scholar] [CrossRef]
- Nasar, F.; Palacios, G.; Gorchakov, R.V.; Guzman, H.; Da Rosa, A.P.; Savji, N.; Popov, V.L.; Sherman, M.B.; Lipkin, W.I.; Tesh, R.B.; et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 14622–14627. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Seymour, R.L.; Kaelber, J.T.; Kim, D.Y.; Leal, G.; Sherman, M.B.; Frolov, I.; Chiu, W.; Weaver, S.C.; Nasar, F. Novel insect-specific eilat virus-based chimeric vaccine candidates provide durable, mono- and multivalent, single-dose protection against lethal alphavirus challenge. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Grieder, F.B.; Davis, B.K.; Zhou, X.D.; Chen, S.J.; Finkelman, F.D.; Gause, W.C. Kinetics of cytokine expression and regulation of host protection following infection with molecularly cloned venezuelan equine encephalitis virus. Virology 1997, 233, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Rosas, C.T.; Paessler, S.; Ni, H.; Osterrieder, N. Protection of mice by equine herpesvirus type 1 based experimental vaccine against lethal venezuelan equine encephalitis virus infection in the absence of neutralizing antibodies. Am. J. Trop Med. Hyg. 2008, 78, 83–92. [Google Scholar] [CrossRef]
- Hu, W.G.; Steigerwald, R.; Kalla, M.; Volkmann, A.; Noll, D.; Nagata, L.P. Protective efficacy of monovalent and trivalent recombinant MVA-based vaccines against three encephalitic alphaviruses. Vaccine 2018, 36, 5194–5203. [Google Scholar] [CrossRef] [PubMed]
- Timm, A.; Enzinger, C.; Felder, E.; Chaplin, P. Genetic stability of recombinant MVA-BN. Vaccine 2006, 24, 4618–4621. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.R.; Short, W.A.; Johnson, A.J.; Bolin, R.A.; Roehrig, J.T. Synthetic peptides of the E2 glycoprotein of venezuelan equine encephalomyelitis virus. II. Antibody to the amino terminus protects animals by limiting viral replication. Virology 1991, 185, 281–290. [Google Scholar] [CrossRef]
- Rico, A.B.; Phillips, A.T.; Schountz, T.; Jarvis, D.L.; Tjalkens, R.B.; Powers, A.M.; Olson, K.E. Venezuelan and western equine encephalitis virus e1 liposome antigen nucleic acid complexes protect mice from lethal challenge with multiple alphaviruses. Virology 2016, 499, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Brooke, C.B.; Deming, D.J.; Whitmore, A.C.; White, L.J.; Johnston, R.E. T cells facilitate recovery from venezuelan equine encephalitis virus-induced encephalomyelitis in the absence of antibody. J. Virol 2010, 84, 4556–4568. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef]
- Saxena, M.; Van, T.T.; Baird, F.J.; Coloe, P.J.; Smooker, P.M. Pre-existing immunity against vaccine vectors--friend or foe? Microbiology 2013, 159, 1–11. [Google Scholar] [CrossRef]
- Riemenschneider, J.; Garrison, A.; Geisbert, J.; Jahrling, P.; Hevey, M.; Negley, D.; Schmaljohn, A.; Lee, J.; Hart, M.K.; Vanderzanden, L.; et al. Comparison of individual and combination DNA vaccines for b. Anthracis, ebola virus, marburg virus and venezuelan equine encephalitis virus. Vaccine 2003, 21, 4071–4080. [Google Scholar] [CrossRef]
- Dupuy, L.C.; Richards, M.J.; Reed, D.S.; Schmaljohn, C.S. Immunogenicity and protective efficacy of a DNA vaccine against venezuelan equine encephalitis virus aerosol challenge in nonhuman primates. Vaccine 2010, 28, 7345–7350. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, L.C.; Richards, M.J.; Ellefsen, B.; Chau, L.; Luxembourg, A.; Hannaman, D.; Livingston, B.D.; Schmaljohn, C.S. A DNA vaccine for venezuelan equine encephalitis virus delivered by intramuscular electroporation elicits high levels of neutralizing antibodies in multiple animal models and provides protective immunity to mice and nonhuman primates. Clin. Vaccine Immunol. 2011, 18, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Hannaman, D.; Dupuy, L.C.; Ellefsen, B.; Schmaljohn, C.S. A phase 1 clinical trial of a DNA vaccine for venezuelan equine encephalitis delivered by intramuscular or intradermal electroporation. Vaccine 2016, 34, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Bounds, C.E.; Terry, F.E.; Moise, L.; Hannaman, D.; Martin, W.D.; De Groot, A.S.; Suschak, J.J.; Dupuy, L.C.; Schmaljohn, C.S. An immunoinformatics-derived DNA vaccine encoding human class II T cell epitopes of ebola virus, sudan virus, and venezuelan equine encephalitis virus is immunogenic in HLA transgenic mice. Hum. Vaccin Immunother. 2017, 13, 2824–2836. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, L.C.; Locher, C.P.; Paidhungat, M.; Richards, M.J.; Lind, C.M.; Bakken, R.; Parker, M.D.; Whalen, R.G.; Schmaljohn, C.S. Directed molecular evolution improves the immunogenicity and protective efficacy of a venezuelan equine encephalitis virus DNA vaccine. Vaccine 2009, 27, 4152–4160. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, I.; Lukashevich, I.S.; Glass, P.; Wang, E.; Weaver, S.; Pushko, P. Novel vaccine against venezuelan equine encephalitis combines advantages of DNA immunization and a live attenuated vaccine. Vaccine 2013, 31, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Konopka, J.L.; Thompson, J.M.; Whitmore, A.C.; Webb, D.L.; Johnston, R.E. Acute infection with venezuelan equine encephalitis virus replicon particles catalyzes a systemic antiviral state and protects from lethal virus challenge. J. Virol. 2009, 83, 12432–12442. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Whitmore, A.C.; Konopka, J.L.; Collier, M.L.; Richmond, E.M.; Davis, N.L.; Staats, H.F.; Johnston, R.E. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. USA 2006, 103, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Nicholson, M.G.; Whitmore, A.C.; Zamora, M.; West, A.; Iwasaki, A.; Staats, H.F.; Johnston, R.E. Nonmucosal alphavirus vaccination stimulates a mucosal inductive environment in the peripheral draining lymph node. J. Immunol. 2008, 181, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Phillpotts, R.J.; O’Brien, L.; Appleton, R.E.; Carr, S.; Bennett, A. Intranasal immunisation with defective adenovirus serotype 5 expressing the venezuelan equine encephalitis virus E2 glycoprotein protects against airborne challenge with virulent virus. Vaccine 2005, 23, 1615–1623. [Google Scholar] [CrossRef]
- Perkins, S.D.; O’Brien, L.M.; Phillpotts, R.J. Boosting with an adenovirus-based vaccine improves protective efficacy against venezuelan equine encephalitis virus following DNA vaccination. Vaccine 2006, 24, 3440–3445. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; O’Brien, L.M.; Phillpotts, R.J.; Perkins, S.D. Improved efficacy of a gene optimised adenovirus-based vaccine for venezuelan equine encephalitis virus. Virol. J. 2009, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.S.; Glass, P.J.; Bakken, R.R.; Barth, J.F.; Lind, C.M.; da Silva, L.; Hart, M.K.; Rayner, J.; Alterson, K.; Custer, M.; et al. Combined alphavirus replicon particle vaccine induces durable and cross-protective immune responses against equine encephalitis viruses. J. Virol. 2014, 88, 12077–12086. [Google Scholar] [CrossRef] [PubMed]
- Phillpotts, R.J. Venezuelan equine encephalitis virus complex-specific monoclonal antibody provides broad protection, in murine models, against airborne challenge with viruses from serogroups I, II and III. Virus Res. 2006, 120, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.D.; Buckley, M.J.; Melanson, V.R.; Glass, P.J.; Norwood, D.; Hart, M.K. Antibody to the e3 glycoprotein protects mice against lethal venezuelan equine encephalitis virus infection. J. Virol. 2010, 84, 12683–12690. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.M.; Underwood-Fowler, C.D.; Goodchild, S.A.; Phelps, A.L.; Phillpotts, R.J. Development of a novel monoclonal antibody with reactivity to a wide range of venezuelan equine encephalitis virus strains. Virol. J. 2009, 6, 206. [Google Scholar] [CrossRef]
- Porta, J.; Jose, J.; Roehrig, J.T.; Blair, C.D.; Kuhn, R.J.; Rossmann, M.G. Locking and blocking the viral landscape of an alphavirus with neutralizing antibodies. J. Virol. 2014, 88, 9616–9623. [Google Scholar] [CrossRef]
- Hu, W.G.; Phelps, A.L.; Jager, S.; Chau, D.; Hu, C.C.; O’Brien, L.M.; Perkins, S.D.; Gates, A.J.; Phillpotts, R.J.; Nagata, L.P. A recombinant humanized monoclonal antibody completely protects mice against lethal challenge with venezuelan equine encephalitis virus. Vaccine 2010, 28, 5558–5564. [Google Scholar] [CrossRef]
- Hunt, A.R.; Bowen, R.A.; Frederickson, S.; Maruyama, T.; Roehrig, J.T.; Blair, C.D. Treatment of mice with human monoclonal antibody 24h after lethal aerosol challenge with virulent venezuelan equine encephalitis virus prevents disease but not infection. Virology 2011, 414, 146–152. [Google Scholar] [CrossRef]
- Hunt, A.R.; Frederickson, S.; Hinkel, C.; Bowdish, K.S.; Roehrig, J.T. A humanized murine monoclonal antibody protects mice either before or after challenge with virulent venezuelan equine encephalomyelitis virus. J. Gen. Virol. 2006, 87, 2467–2476. [Google Scholar] [CrossRef]
- Braid, L.R.; Hu, W.G.; Davies, J.E.; Nagata, L.P. Engineered mesenchymal cells improve passive immune protection against lethal venezuelan equine encephalitis virus exposure. Stem Cells Transl. Med. 2016, 5, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, J.S. Usage of human mesenchymal stem cells in cell-based therapy: Advantages and disadvantages. Dev. Reprod. 2017, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiang, W.; Yi, D.Y.; Xue, B.Z.; Wen, W.W.; Abdelmaksoud, A.; Xiong, N.X.; Jiang, X.B.; Zhao, H.Y.; Fu, P. Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res. Ther. 2018, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.G.; Chau, D.; Wu, J.; Jager, S.; Nagata, L.P. Humanization and mammalian expression of a murine monoclonal antibody against venezuelan equine encephalitis virus. Vaccine 2007, 25, 3210–3214. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, S.A.; O’Brien, L.M.; Steven, J.; Muller, M.R.; Lanning, O.J.; Logue, C.H.; D’Elia, R.V.; Phillpotts, R.J.; Perkins, S.D. A humanised murine monoclonal antibody with broad serogroup specificity protects mice from challenge with venezuelan equine encephalitis virus. Antiviral Res. 2011, 90, 1–8. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.M.; Goodchild, S.A.; Phillpotts, R.J.; Perkins, S.D. A humanized murine monoclonal antibody protects mice from venezuelan equine encephalitis virus, everglades virus and mucambo virus when administered up to 48 h after airborne challenge. Virology 2012, 462, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Paessler, S.; Yun, N.E.; Judy, B.M.; Dziuba, N.; Zacks, M.A.; Grund, A.H.; Frolov, I.; Campbell, G.A.; Weaver, S.C.; Estes, D.M. Alpha-beta t cells provide protection against lethal encephalitis in the murine model of VEEV infection. Virology 2007, 367, 307–323. [Google Scholar] [CrossRef]
- Taylor, K.; Kolokoltsova, O.; Ronca, S.E.; Estes, M.; Paessler, S. Live, attenuated venezuelan equine encephalitis virus vaccine (TC83) causes persistent brain infection in mice with non-functional alphabeta T-cells. Front. Microbiol. 2017, 8, 81. [Google Scholar] [CrossRef]
- Yun, N.E.; Peng, B.H.; Bertke, A.S.; Borisevich, V.; Smith, J.K.; Smith, J.N.; Poussard, A.L.; Salazar, M.; Judy, B.M.; Zacks, M.A.; et al. Cd4+ T cells provide protection against acute lethal encephalitis caused by venezuelan equine encephalitis virus. Vaccine 2009, 27, 4064–4073. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VEEV Strains | Mutation | Phenotype | Reference |
|---|---|---|---|
| TrD | Parent strain (E1 clone) | Virulent wild-type | Kinney et al. [21] |
| V3000 | Full-length clone of TrD Envelope protein (E) 2 170 silent mutation removing SmaI E2 239 (Asn to Ile) | Virulent | Grieder et al. [15] |
| TC-83 | 5’ noncoding nt 3 G to A nsp3 codon 260 (Ser to Thr) E2 codon 7 (Lys to Asn) E2 codon 85 (His to Tyr) E2 codon 120 (Thr to Arg) E2 codon 192 (Val to Asp) E2 codon 278 (none) E2 codon 296 (Thr to Ile) E1 codon 161 (Leu to Ile) E1 codon 211 (none) 3’ noncoding nt 11,405 UU to U | Attenuated (IND vaccine) | Kinney et al. [21] |
| V3010 | E2 codon 76 (Glu to Lys) | Attenuated | Davis et al. [18] |
| V3034 | E1 codon 272 (Ala to Thr) | Attenuated | Johnston and Smith [22] Davis et al. [18] Grieder et al. [15] |
| V3032 | E2 codon 209 (Glu to Lys) | Attenuated | Johnston and Smith [22] Davis et al. [18] Grieder et al. [15] |
| V3014 | E2 codon 209 (Glu to Lys) E1 codon 272 (Ala to Thr) E2 codon 239 (Ile to Asn) | Attenuated | Davis et al. [18] Grieder et al. [15] |
| V3533 | E2 codon 76 (Glu to Lys) E2 codon 116 (Lys to Glu) | Attenuated | Aronson et al. [15] |
| V3526 | E3 Δ (56-59) (Furin site cleavage)E1 codon 253 (Phe to Ser) | Attenuated | Davis et al. [23] |
| Vaccine Type | Strain/Antigen | Immunity | Status | Reference |
|---|---|---|---|---|
| Live-attenuated | TC-83 | Sterile | IND | Alevizato et al. [123] Pittman et al. [123] |
| V3526 | Sterile | Phase I | Pratt et al. [128] Holley et al. [132] | |
| Inactivated | Formalin inactivated TrD | Sterile | Equine vaccine (discontinued) | Cole et al. [134] |
| Formalin inactivated C84 (TC-83) * | Poor immunogenicity | IND (Booster) Veterinary vaccine | Edelman et al. [137] | |
| INA-inactivated V3000 and V3526 | Sterile | Pre-clinical | Sharma et al. [133] Gupta et al. [142] | |
| Gamma-irradiated V3526 | Sterile | Pre-clinical | Fine et al. [139] Gayen et al. [145] | |
| Chimera | VEEV/mutSG/IRES/1 (TC-83) | Sterile | Pre-clinical | Volkova et al. [147] |
| VEEV/IRES/C (TC-83) | Sterile | Pre-clinical | Guerbois et al. [148] | |
| VEEV/IRESv1 (68U201) VEEV/IRESv2 (68U201) | Sterile | Pre-clinical | Rossi et al. [149] | |
| VEEV/IRES-Cm (TC-83) | Sterile | Pre-clinical | Atasheva et al. [150] | |
| SINV/VEEV (TC-83) | Non-sterile | Pre-clinical | Paessler et al. [151] | |
| SINV/VEEV (TrD) | Non-sterile | Pre-clinical | Paessler et al. [152] | |
| SINV/VEEV (ZPC738) | Non-sterile | Pre-clinical | Paessler et al. [152] | |
| EILV/VEEV (TC-83) | Sterile | Pre-clinical | Erasmus et al. [154] | |
| EHV-1/VEEV (TC-83) | Sterile | Pre-clinical | Rosas et al. [156] | |
| MVA-BN/VEEV (TrD) | Sterile | Pre-clinical | Hu et al. [157] | |
| Subunit | pWRG7077/VEEV (TrD structural genes) | Non-sterile | Pre-clinical | Riemenschneider et al. [164] Dupuy et al. [165] |
| pWRG7077/VEEV (TrD envelope genes) | Sterile | Phase I | Dupuy et al. [166] Hannaman et al. [167] | |
| pWRG7077/VEEV (TrD Structural genes with T-cell epitope optimized) | Sterile | Pre-clinical | Bounds et al. [168] | |
| pWRG7077/VEEV (TrD and IE E2) | Sterile | Pre-clinical | Dupuy et al. [169] | |
| pcDNA3.1/VEEV (TC-83) | Non-sterile | Pre-clinical | Tretyakova et al. [170] | |
| LANAC (TrD E1) | Sterile | Pre-clinical | Rico et al. [160] | |
| Replicon particles | VEEV VRP (V3000 E2 and E1) | Non-sterile (6h before challenge) | Pre-clinical | Konopka et al. [171] |
| Rad/VEEV (TrD E2) | Sterile | Pre-clinical | Phillpotts et al. [174] Williams et al. [176] | |
| Rad/VEEV (TC-83 E2) | Sterile | Pre-clinical (Booster) | Perkins et al. [175] | |
| V3014 VRP (V3014 PE2 | Sterile | Pre-clinical | Reed et al. [177] | |
| Passive | 1A4A-1 Hu1A4A1IgG1-2A | Sterile (Prophylactic) Non-sterile (Therapeutic) | Pre-clinical | Hu et al. [188] Hu et al. [182] Phillpotts et al. [178] |
| Hu Mab F5nIgG | Non-sterile | Pre-clinical | Hunt et al. [183] | |
| Hu1A3B7 (E2) | post infection | Pre-clinical | Goodchild et al. [189] O’Brien et al. [190] | |
| 13D4 (Anti-E3) | Non-sterile | Pre-clinical | Parker et al. [179] | |
| CUF37-2a (Anti-E2) | Non-sterile | Pre-clinical | O’Brien et al. [180] | |
| 3B4C-4, Hu Mab Hy4-26C | Non-sterile | Pre-clinical | Hunt et al. [184] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Knollmann-Ritschel, B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses 2019, 11, 164. https://doi.org/10.3390/v11020164
Sharma A, Knollmann-Ritschel B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses. 2019; 11(2):164. https://doi.org/10.3390/v11020164
Chicago/Turabian StyleSharma, Anuj, and Barbara Knollmann-Ritschel. 2019. "Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development" Viruses 11, no. 2: 164. https://doi.org/10.3390/v11020164
APA StyleSharma, A., & Knollmann-Ritschel, B. (2019). Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses, 11(2), 164. https://doi.org/10.3390/v11020164