Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections


Abstract
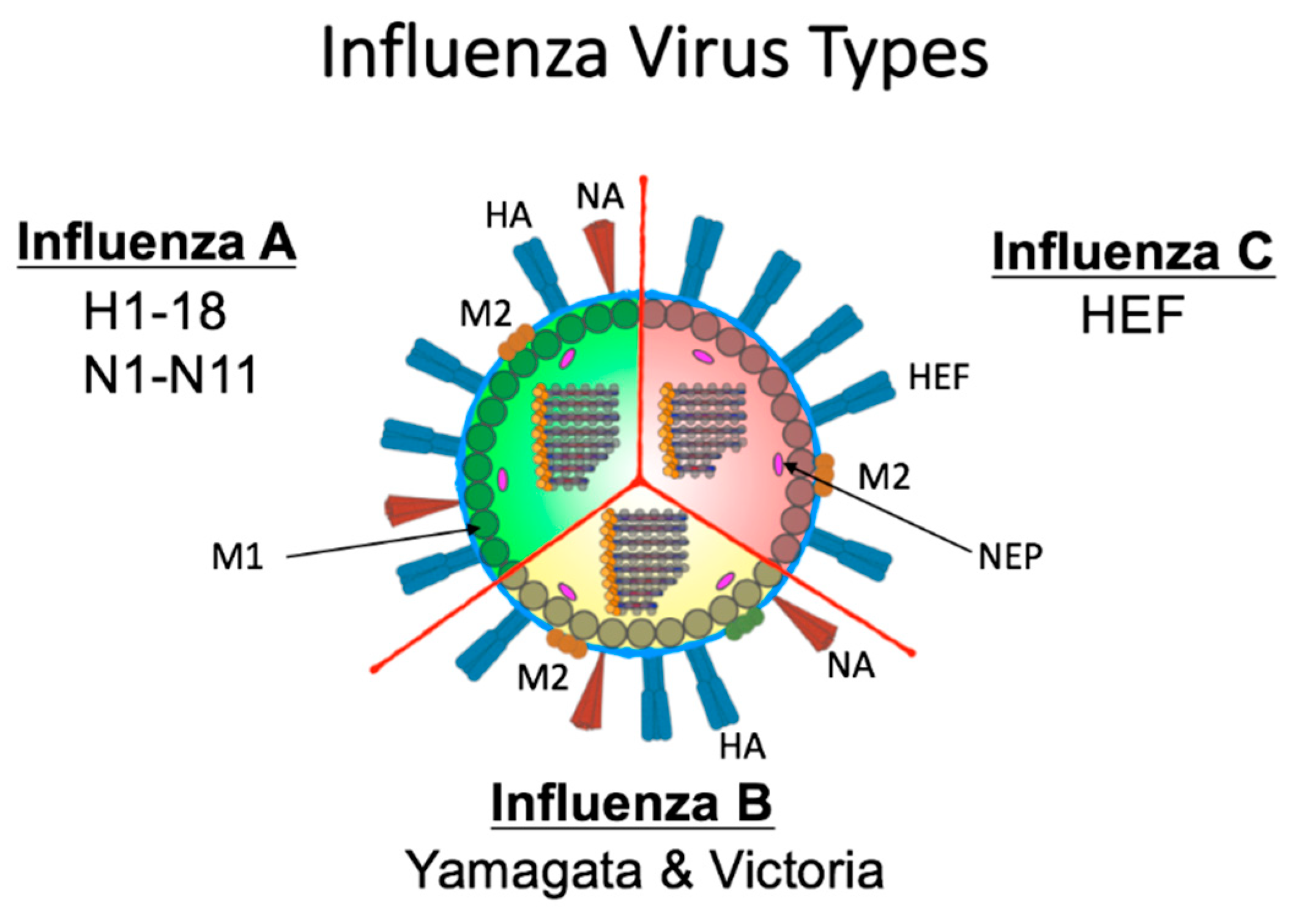
1. Introduction: Influenza Viruses are the Archetype Emerging and Re-Emerging Viruses
2. Influenza Virus Biology Enables Frequent Antigenic Change
3. Host Immune Responses to Primary Influenza Virus Infection
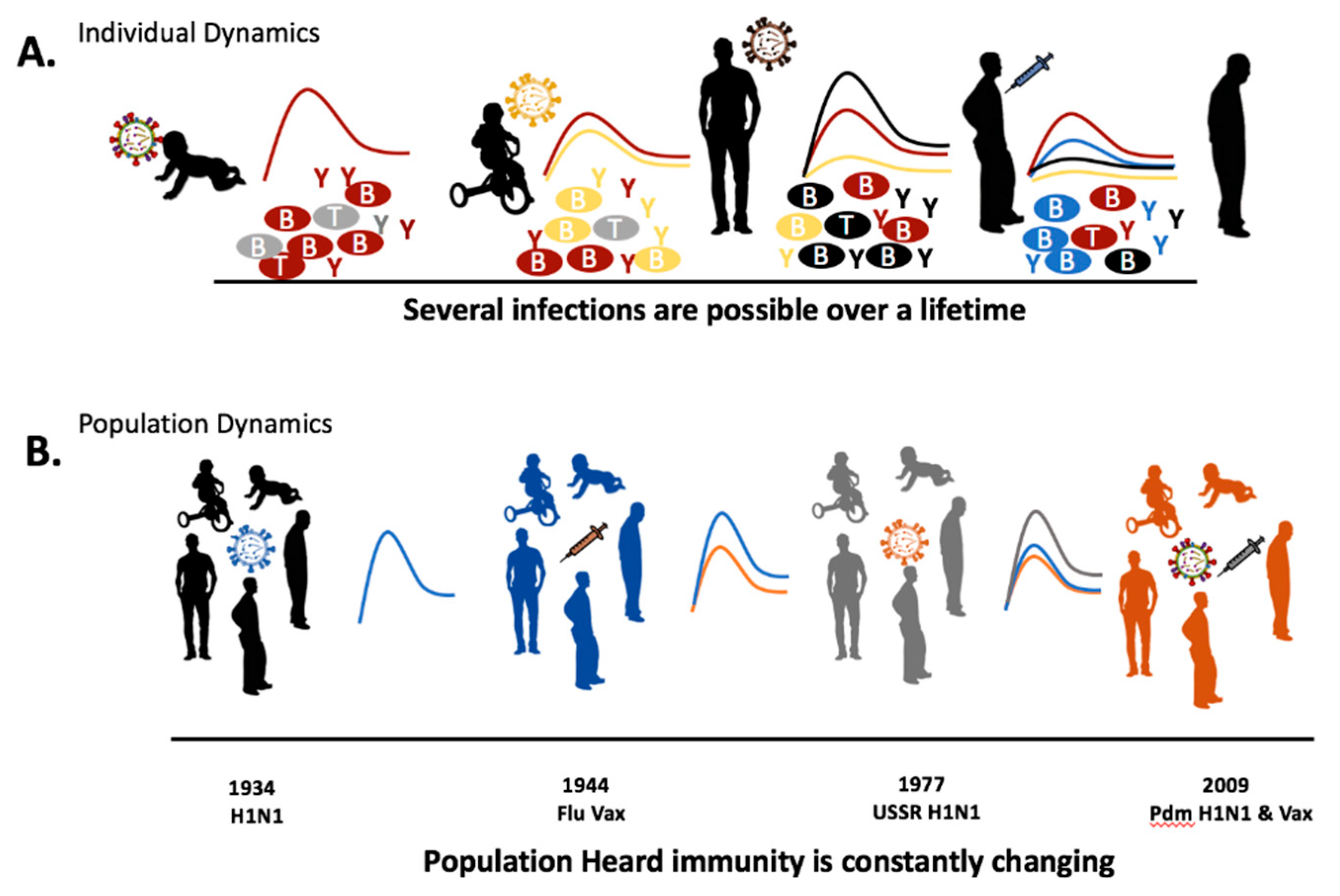
4. Recurrent Infection and Immunological Memory
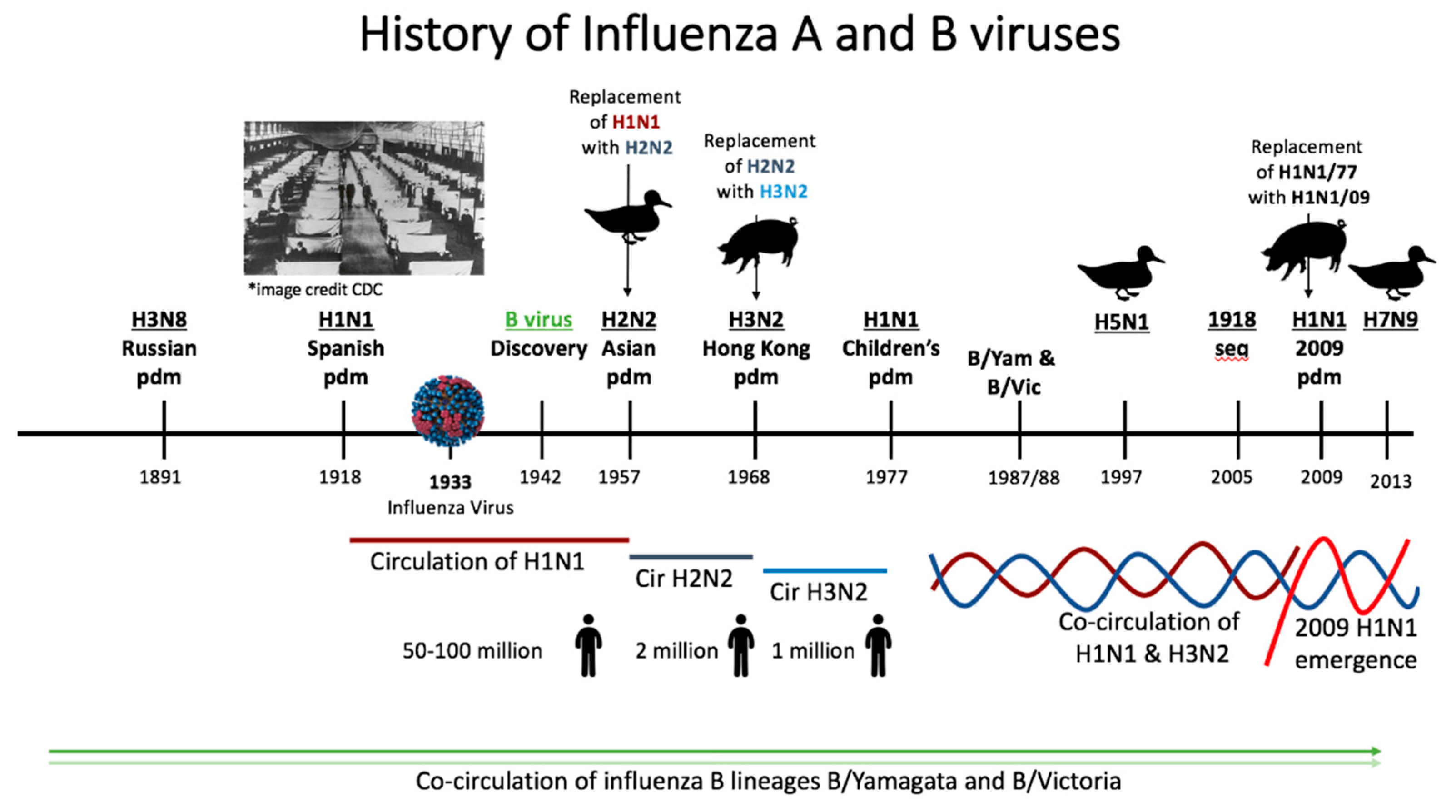
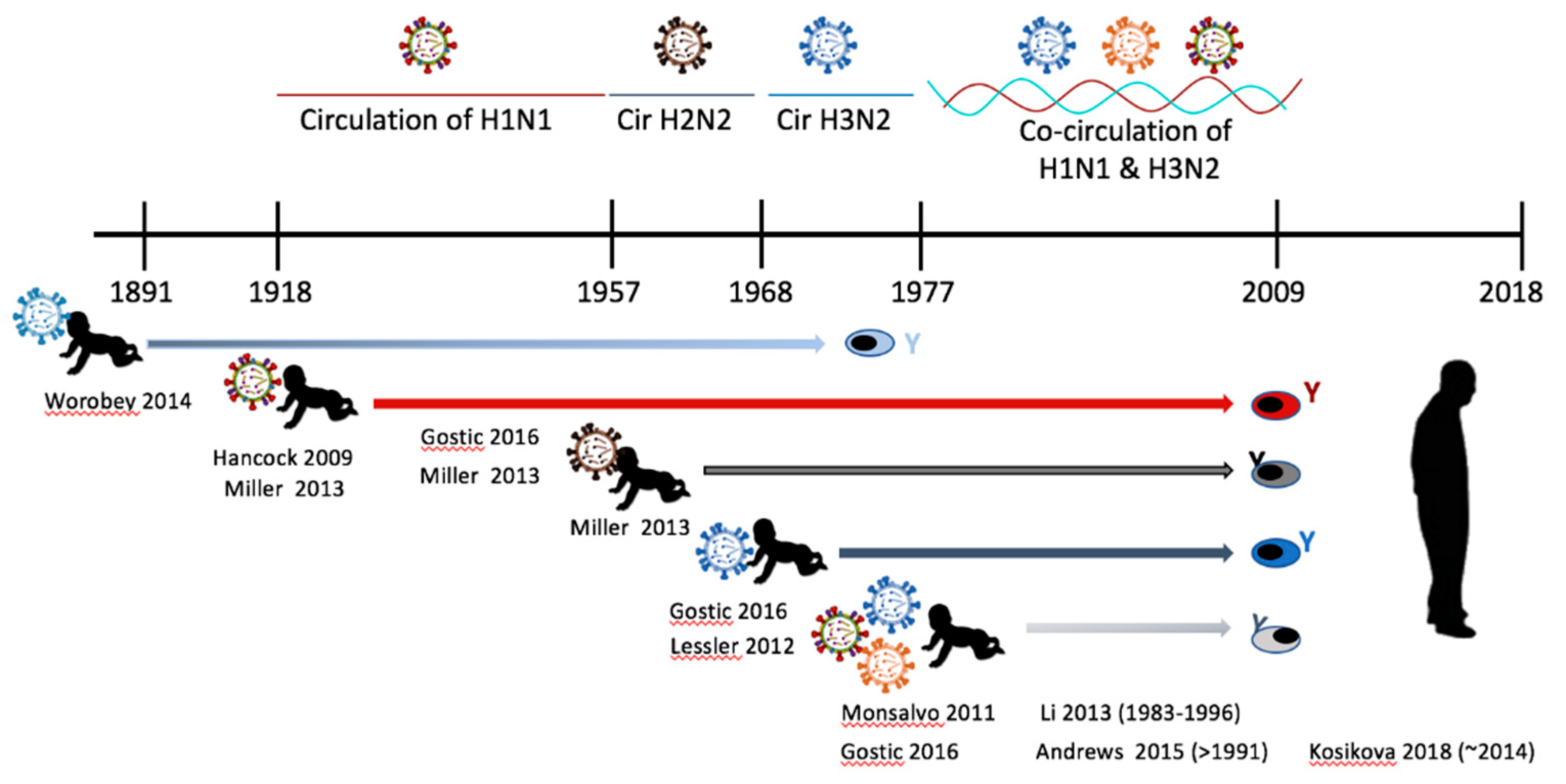
5. Epidemiological History of Influenza Virus Circulation Mirrors the Human Influenza Background
6. Experimental Animal and Human Studies of Preimmunity
7. Vaccination Preimmunity May Influence Future Vaccination Outcome
8. Future Directions
9. Concluding Statement
Funding
Acknowledgments
Conflicts of Interest
References
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Saunders-Hastings, P.R.; Krewski, D. Reviewing the History of Pandemic Influenza: Understanding Patterns of Emergence and Transmission. Pathogens 2016, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Barberis, I.; Myles, P.; Ault, S.K.; Bragazzi, N.L.; Martini, M. History and evolution of influenza control through vaccination: From the first monovalent vaccine to universal vaccines. J. Prev. Med. Hyg. 2016, 57, E115–E120. [Google Scholar] [PubMed]
- Neu, K.E.; Henry Dunand, C.J.; Wilson, P.C. Heads, stalks and everything else: How can antibodies eradicate influenza as a human disease? Curr. Opin. Immunol. 2016, 42, 48–55. [Google Scholar] [CrossRef] [PubMed]
- WHO. Biologicals: Influenza. Available online: https://www.who.int/biologicals/vaccines/influenza/en/ (accessed on December 2018).
- Hay, A.J.; Gregory, V.; Douglas, A.R.; Lin, Y.P. The evolution of human influenza viruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Francis, T.J. On the Doctrine of Original Antigenic Sin. Proc. Am. Philos. Soc. 1960, 104, 572–578. [Google Scholar]
- Fonville, J.M.; Wilks, S.H.; James, S.L.; Fox, A.; Ventresca, M.; Aban, M.; Xue, L.; Jones, T.C.; Le, N.M.H.; Pham, Q.T.; et al. Antibody landscapes after influenza virus infection or vaccination. Science 2014, 346, 996–1000. [Google Scholar] [CrossRef]
- Monto, A.S.; Malosh, R.E.; Petrie, J.G.; Martin, E.T. The Doctrine of Original Antigenic Sin: Separating Good From Evil. J. Infect. Dis. 2017, 215, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Lessler, J.; Riley, S.; Read, J.M.; Wang, S.; Zhu, H.; Smith, G.J.; Guan, Y.; Jiang, C.Q.; Cummings, D.A. Evidence for antigenic seniority in influenza A (H3N2) antibody responses in southern China. PLoS Pathog. 2012, 8, e1002802. [Google Scholar] [CrossRef]
- Skowronski, D.M.; Chambers, C.; De Serres, G.; Sabaiduc, S.; Winter, A.L.; Dickinson, J.A.; Gubbay, J.B.; Fonseca, K.; Drews, S.J.; Charest, H.; et al. Serial Vaccination and the Antigenic Distance Hypothesis: Effects on Influenza Vaccine Effectiveness During A(H3N2) Epidemics in Canada, 2010–2011 to 2014–2015. J. Infect. Dis. 2017, 215, 1059–1099. [Google Scholar] [CrossRef]
- Miller, M.S.; Gardner, T.J.; Krammer, F.; Aguado, L.C.; Tortorella, D.; Basler, C.F.; Palese, P. Neutralizing antibodies against previously encountered influenza virus strains increase over time: A longitudinal analysis. Sci. Transl. Med. 2013, 5, 198ra107. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Banner, D.; Kelvin, A.A.; Huang, S.S.; Paige, C.J.; Corfe, S.A.; Kane, K.P.; Bleackley, R.C.; Rowe, T.; Leon, A.J.; et al. Seasonal H1N1 influenza virus infection induces cross-protective pandemic H1N1 virus immunity through a CD8-independent, B cell-dependent mechanism. J. Virol. 2012, 86, 2229–2238. [Google Scholar] [CrossRef]
- Andrews, S.F.; Huang, Y.; Kaur, K.; Popova, L.I.; Ho, I.Y.; Pauli, N.T.; Henry Dunand, C.J.; Taylor, W.M.; Lim, S.; Huang, M.; et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 2015, 7, 316ra192. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Gostic, K.M.; Ambrose, M.; Worobey, M.; Lloyd-Smith, J.O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 2016, 354, 722–726. [Google Scholar] [CrossRef]
- Henry, C.; Palm, A.E.; Krammer, F.; Wilson, P.C. From Original Antigenic Sin to the Universal Influenza Virus Vaccine. Trends Immunol. 2018, 39, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Hancock, K.; Veguilla, V.; Lu, X.; Zhong, W.; Butler, E.N.; Sun, H.; Liu, F.; Dong, L.; DeVos, J.R.; Gargiullo, P.M.; et al. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N. Engl. J. Med. 2009, 361, 1945–1952. [Google Scholar] [CrossRef]
- Cobey, S.; Hensley, S.E. Immune history and influenza virus susceptibility. Curr. Opin. Virol. 2017, 22, 105–111. [Google Scholar] [CrossRef]
- Lewnard, J.A.; Cobey, S. Immune History and Influenza Vaccine Effectiveness. Vaccines (Basel) 2018, 6, 28. [Google Scholar] [CrossRef]
- Ducatez, M.F.; Pelletier, C.; Meyer, G. Influenza D virus in cattle, France, 2011–2014. Emerg. Infect. Dis. 2015, 21, 368–371. [Google Scholar] [CrossRef]
- WHO. Influenza Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/2003/fs211/en/ (accessed on December 2018).
- Herrler, G.; Klenk, H.D. Structure and function of the HEF glycoprotein of influenza C virus. Adv. Virus Res. 1991, 40, 213–234. [Google Scholar] [PubMed]
- Lofgren, E.; Fefferman, N.H.; Naumov, Y.N.; Gorski, J.; Naumova, E.N. Influenza seasonality: Underlying causes and modeling theories. J. Virol. 2007, 81, 5429–5436. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Tumpey, T.M.; Basler, C.F.; Aguilar, P.V.; Zeng, H.; Solorzano, A.; Swayne, D.E.; Cox, N.J.; Katz, J.M.; Taubenberger, J.K.; Palese, P.; et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 2005, 310, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Lourens, R.M.; Wang, R.; Jin, G.; Fanning, T.G. Characterization of the 1918 influenza virus polymerase genes. Nature 2005, 437, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Palese, P.; Wang, T.T. Why do influenza virus subtypes die out? A hypothesis. MBio 2011, 2, e00150-11. [Google Scholar] [CrossRef]
- Krammer, F.; Pica, N.; Hai, R.; Tan, G.S.; Palese, P. Hemagglutinin Stalk-Reactive Antibodies Are Boosted following Sequential Infection with Seasonal and Pandemic H1N1 Influenza Virus in Mice. J. Virol. 2012, 86, 10302–10307. [Google Scholar] [CrossRef]
- Pica, N.; Hai, R.; Krammer, F.; Wang, T.T.; Maamary, J.; Eggink, D.; Tan, G.S.; Krause, J.C.; Moran, T.; Stein, C.R.; et al. Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 2573–2578. [Google Scholar] [CrossRef]
- Kirchenbaum, G.A.; Carter, D.M.; Ross, T.M. Sequential Infection in Ferrets with Antigenically Distinct Seasonal H1N1 Influenza Viruses Boosts Hemagglutinin Stalk-Specific Antibodies. J. Virol. 2015, 90, 1116–1128. [Google Scholar] [CrossRef]
- Paquette, S.G.; Huang, S.S.; Banner, D.; Xu, L.; Leomicronn, A.; Kelvin, A.A.; Kelvin, D.J. Impaired heterologous immunity in aged ferrets during sequential influenza A H1N1 infection. Virology 2014, 464–465, 177–183. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Banner, D.; Paquette, S.G.; Leon, A.J.; Kelvin, A.A.; Kelvin, D.J. Pathogenic influenza B virus in the ferret model establishes lower respiratory tract infection. J. Gen. Virol. 2014, 95, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R.; Morick, D.; de Mutsert, G.; Osinga, N.; Bestebroer, T.; van der Vliet, S.; Smits, S.L.; Kuiken, T.; Rimmelzwaan, G.F.; Fouchier, R.A.; et al. Recurring influenza B virus infections in seals. Emerg. Infect. Dis. 2013, 19, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Kanegae, Y.; Sugita, S.; Endo, A.; Ishida, M.; Senya, S.; Osako, K.; Nerome, K.; Oya, A. Evolutionary pattern of the hemagglutinin gene of influenza B viruses isolated in Japan: Cocirculating lineages in the same epidemic season. J. Virol. 1990, 64, 2860–2865. [Google Scholar] [PubMed]
- Shcherbinin, D.N.; Alekseeva, S.V.; Shmarov, M.M.; Smirnov, Y.A.; Naroditskiy, B.S.; Gintsburg, A.L. The Analysis of B-Cell Epitopes of Influenza Virus Hemagglutinin. Acta Nat. 2016, 8, 13–20. [Google Scholar]
- Tan, G.S.; Lee, P.S.; Hoffman, R.M.; Mazel-Sanchez, B.; Krammer, F.; Leon, P.E.; Ward, A.B.; Wilson, I.A.; Palese, P. Characterization of a broadly neutralizing monoclonal antibody that targets the fusion domain of group 2 influenza A virus hemagglutinin. J. Virol. 2014, 88, 13580–13592. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, Y.M. Influenza virus antigenic variation, host antibody production and new approach to control epidemics. Virol. J. 2009, 6, 30. [Google Scholar] [CrossRef]
- Oslund, K.L.; Baumgarth, N. Influenza-induced innate immunity: Regulators of viral replication, respiratory tract pathology & adaptive immunity. Future Virol. 2011, 6, 951–962. [Google Scholar]
- Manicassamy, B.; Manicassamy, S.; Belicha-Villanueva, A.; Pisanelli, G.; Pulendran, B.; Garcia-Sastre, A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. USA 2010, 107, 11531–11536. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, N.; Sato, Y.; Katano, H.; Hasegawa, H.; Kumasaka, T.; Hata, S.; Tanaka, S.; Amano, T.; Kasai, T.; Chong, J.M.; et al. Histopathological and immunohistochemical findings of 20 autopsy cases with 2009 H1N1 virus infection. Mod. Pathol. 2012, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Dong, Z.; Li, F.; Meng, W.; Feng, L.; Niu, X.; Li, C.; Luo, Q.; Li, Z.; Sun, C.; et al. Visualizing influenza virus infection in living mice. Nat. Commun. 2013, 4, 2369. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Maddur, M.S. Innate immune sensing and response to influenza. Curr. Top. Microbiol. Immunol. 2015, 386, 23–71. [Google Scholar] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef]
- Waithman, J.; Mintern, J.D. Dendritic cells and influenza A virus infection. Virulence 2012, 3, 603–608. [Google Scholar] [CrossRef]
- Leon, A.J.; Banner, D.; Xu, L.; Ran, L.; Peng, Z.; Yi, K.; Chen, C.; Xu, F.; Huang, J.; Zhao, Z.; et al. Sequencing, annotation, and characterization of the influenza ferret infectome. J. Virol. 2013, 87, 1957–1966. [Google Scholar] [CrossRef]
- Rowe, T.; Leon, A.J.; Crevar, C.J.; Carter, D.M.; Xu, L.; Ran, L.; Fang, Y.; Cameron, C.M.; Cameron, M.J.; Banner, D.; et al. Modeling host responses in ferrets during A/California/07/2009 influenza infection. Virology 2010, 401, 257–265. [Google Scholar] [CrossRef]
- Sun, J.; Braciale, T.J. Role of T cell immunity in recovery from influenza virus infection. Curr. Opin. Virol. 2013, 3, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Grant, E.J.; Quinones-Parra, S.M.; Clemens, E.B.; Kedzierska, K. Human influenza viruses and CD8(+) T cell responses. Curr. Opin. Virol. 2016, 16, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.M.; Kim, T.S.; Sun, J.; Braciale, T.J. Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J. Exp. Med. 2011, 208, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Dolfi, D.V.; Duttagupta, P.A.; Boesteanu, A.C.; Mueller, Y.M.; Oliai, C.H.; Borowski, A.B.; Katsikis, P.D. Dendritic cells and CD28 costimulation are required to sustain virus-specific CD8+ T cell responses during the effector phase in vivo. J. Immunol. 2011, 186, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Buck, A.; Curtis, J.D.; Dibble, J.P.; Huston, G.; Tighe, M.; Hamada, H.; Sell, S.; Dutton, R.W.; et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J. Immunol. 2009, 182, 7353–7363. [Google Scholar] [CrossRef]
- Smith, K.G.; Light, A.; Nossal, G.J.; Tarlinton, D.M. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997, 16, 2996–3006. [Google Scholar] [CrossRef]
- Neu, K.E.; Wilson, P.C. Taking the Broad View on B Cell Affinity Maturation. Immunity 2016, 44, 518–520. [Google Scholar] [CrossRef]
- Krammer, F.; Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 2013, 3, 521–530. [Google Scholar] [CrossRef]
- Bui, H.H.; Peters, B.; Assarsson, E.; Mbawuike, I.; Sette, A. Ab and T cell epitopes of influenza A virus, knowledge and opportunities. Proc. Natl. Acad. Sci. USA 2007, 104, 246–251. [Google Scholar] [CrossRef]
- Ellebedy, A.H.; Webby, R.J. Influenza vaccines. Vaccine 2009, 27, D65–D68. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S. Seasonal influenza and vaccination coverage. Vaccine 2010, 28, D33–D44. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Tarlinton, D.M. Memory B cells: Effectors of long-lived immune responses. Eur. J. Immunol. 2009, 39, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Koopman, G.; Mooij, P.; Dekking, L.; Mortier, D.; Nieuwenhuis, I.G.; van Heteren, M.; Kuipers, H.; Remarque, E.J.; Radosevic, K.; Bogers, W.M. Correlation between Virus Replication and Antibody Responses in Macaques following Infection with Pandemic Influenza A Virus. J. Virol. 2016, 90, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Immune memory redefined: Characterizing the longevity of natural killer cells. Immunol. Rev. 2010, 236, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Ellebedy, A.H.; Wrammert, J.; Ahmed, R. B cell responses to influenza infection and vaccination. Curr. Top. Microbiol. Immunol. 2015, 386, 381–398. [Google Scholar]
- Lipsitch, M.; Viboud, C. Influenza seasonality: Lifting the fog. Proc. Natl. Acad. Sci. USA 2009, 106, 3645–3646. [Google Scholar] [CrossRef]
- Smith, W.; Andrewes, C.H.; Laidlaw, P. A virus isolated from influenza patients. Lancet 1933, 225, 66–68. [Google Scholar] [CrossRef]
- Yang, J.R.; Huang, Y.P.; Chang, F.Y.; Hsu, L.C.; Lin, Y.C.; Huang, H.Y.; Wu, F.T.; Wu, H.S.; Liu, M.T. Phylogenetic and evolutionary history of influenza B viruses, which caused a large epidemic in 2011–2012, Taiwan. PLoS ONE 2012, 7, e47179. [Google Scholar] [CrossRef]
- Hannoun, C. The evolving history of influenza viruses and influenza vaccines. Expert Rev. Vacc. 2013, 12, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Janczewski, T.A.; Fanning, T.G. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos. Trans. R Soc. Lond. B Biol. Sci. 2001, 356, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Han, G.Z.; Rambaut, A. Genesis and pathogenesis of the 1918 pandemic H1N1 influenza A virus. Proc. Natl. Acad. Sci. USA 2014, 111, 8107–8112. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Krafft, A.E.; Bijwaard, K.E.; Fanning, T.G. Initial genetic characterization of the 1918 "Spanish" influenza virus. Science 1997, 275, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.; Bahl, J.; Vijaykrishna, D.; Zhang, J.; Poon, L.L.; Chen, H.; Webster, R.G.; Peiris, J.S.; Guan, Y. Dating the emergence of pandemic influenza viruses. Proc. Natl. Acad. Sci. USA 2009, 106, 11709–11712. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O. The re-emergence of H1N1 influenza virus in 1977: A cautionary tale for estimating divergence times using biologically unrealistic sampling dates. PLoS ONE 2010, 5, e11184. [Google Scholar] [CrossRef] [PubMed]
- CDC. Estimated Influenza Illnesses, Medical Visits, Hospitalizations, and Deaths Averted by Vaccination in the United States. Available online: https://www.cdc.gov/flu/about/disease/burden-averted-vaccination.htm. (accessed on December 2018).
- Shinde, V.; Bridges, C.B.; Uyeki, T.M.; Shu, B.; Balish, A.; Xu, X.; Lindstrom, S.; Gubareva, L.V.; Deyde, V.; Garten, R.J.; et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 2009, 360, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Qin, Y.; Cowling, B.J.; Ren, X.; Wardrop, N.A.; Gilbert, M.; Tsang, T.K.; Wu, P.; Feng, L.; Jiang, H.; et al. Global epidemiology of avian influenza A H5N1 virus infection in humans, 1997–2015: A systematic review of individual case data. Lancet Infect. Dis. 2016, 16, e108–e118. [Google Scholar] [CrossRef]
- Subbarao, K.; Klimov, A.; Katz, J.; Regnery, H.; Lim, W.; Hall, H.; Perdue, M.; Swayne, D.; Bender, C.; Huang, J.; et al. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 1998, 279, 393–396. [Google Scholar] [CrossRef]
- Khurana, S.; Chung, K.Y.; Coyle, E.M.; Meijer, A.; Golding, H. Antigenic Fingerprinting of Antibody Response in Humans following Exposure to Highly Pathogenic H7N7 Avian Influenza Virus: Evidence for Anti-PA-X Antibodies. J. Virol. 2016, 90, 9383–9393. [Google Scholar] [CrossRef]
- Peiris, M.; Yuen, K.Y.; Leung, C.W.; Chan, K.H.; Ip, P.L.; Lai, R.W.; Orr, W.K.; Shortridge, K.F. Human infection with influenza H9N2. Lancet 1999, 354, 916–917. [Google Scholar] [CrossRef]
- CDC. Human Infection with Highly Pathogenic A(H7N7) Avian Influenza Virus, Italy, 2013. Emerg. Infect. Dis. 2014, 20, 1745. [Google Scholar]
- Xin, L.; Bai, T.; Zhou, J.F.; Chen, Y.K.; Li, X.D.; Zhu, W.F.; Li, Y.; Tang, J.; Chen, T.; Qin, K.; et al. Seropositivity for Avian Influenza H6 Virus among Humans, China. Emerg. Infect. Dis. 2015, 21, 1267. [Google Scholar] [CrossRef] [PubMed]
- Ellebedy, A.H. Immunizing the Immune: Can We Overcome Influenza’s Most Formidable Challenge? Vaccines (Basel) 2018, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Linderman, S.L.; Hensley, S.E. Antibodies with ‘Original Antigenic Sin’ Properties Are Valuable Components of Secondary Immune Responses to Influenza Viruses. PLoS Pathog. 2016, 12, e1005806. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Huang, C.T.; Lin, C.Y.; Chen, T.C.; Lin, Y.C.; Chang, C.S.; He, Y.C. Sterilizing immunity to influenza virus infection requires local antigen-specific T cell response in the lungs. Sci. Rep. 2016, 6, 32973. [Google Scholar] [CrossRef] [PubMed]
- Ellebedy, A.H.; Ahmed, R. Re-engaging cross-reactive memory B cells: The influenza puzzle. Front. Immunol. 2012, 3, 53. [Google Scholar] [CrossRef]
- O’Neill, E.; Krauss, S.L.; Riberdy, J.M.; Webster, R.G.; Woodland, D.L. Heterologous protection against lethal A/HongKong/156/97 (H5N1) influenza virus infection in C57BL/6 mice. J. Gen. Virol. 2000, 81, 2689–2696. [Google Scholar] [CrossRef]
- LaMere, M.W.; Lam, H.T.; Moquin, A.; Haynes, L.; Lund, F.E.; Randall, T.D.; Kaminski, D.A. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J. Immunol. 2011, 186, 4331–4339. [Google Scholar] [CrossRef]
- Laurie, K.L.; Carolan, L.A.; Middleton, D.; Lowther, S.; Kelso, A.; Barr, I.G. Multiple infections with seasonal influenza A virus induce cross-protective immunity against A(H1N1) pandemic influenza virus in a ferret model. J. Infect. Dis. 2010, 202, 1011–1020. [Google Scholar] [CrossRef]
- Li, Y.; Myers, J.L.; Bostick, D.L.; Sullivan, C.B.; Madara, J.; Linderman, S.L.; Liu, Q.; Carter, D.M.; Wrammert, J.; Esposito, S.; et al. Immune history shapes specificity of pandemic H1N1 influenza antibody responses. J. Exp. Med. 2013, 210, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Kosikova, M.; Li, L.; Radvak, P.; Ye, Z.; Wan, X.F.; Xie, H. Imprinting of Repeated Influenza A/H3 Exposures on Antibody Quantity and Antibody Quality: Implications for Seasonal Vaccine Strain Selection and Vaccine Performance. Clin. Infect. Dis. 2018, 67, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Marini, R.P. Biology and Diseases of the Ferret, 3rd ed.; WILEY Blackwell: Hoboken, NJ, USA, 2014. [Google Scholar]
- Kallewaard, N.L.; Corti, D.; Collins, P.J.; Neu, U.; McAuliffe, J.M.; Benjamin, E.; Wachter-Rosati, L.; Palmer-Hill, F.J.; Yuan, A.Q.; Walker, P.A.; et al. Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell 2016, 166, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Nachbagauer, R.; Wohlbold, T.J.; Hirsh, A.; Hai, R.; Sjursen, H.; Palese, P.; Cox, R.J.; Krammer, F. Induction of broadly reactive anti-hemagglutinin stalk antibodies by an H5N1 vaccine in humans. J. Virol. 2014, 88, 13260–13268. [Google Scholar] [CrossRef] [PubMed]
- Nachbagauer, R.; Miller, M.S.; Hai, R.; Ryder, A.B.; Rose, J.K.; Palese, P.; Garcia-Sastre, A.; Krammer, F.; Albrecht, R.A. Hemagglutinin Stalk Immunity Reduces Influenza Virus Replication and Transmission in Ferrets. J. Virol. 2015, 90, 3268–3273. [Google Scholar] [CrossRef] [PubMed]
- Monsalvo, A.C.; Batalle, J.P.; Lopez, M.F.; Krause, J.C.; Klemenc, J.; Hernandez, J.Z.; Maskin, B.; Bugna, J.; Rubinstein, C.; Aguilar, L.; et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 2011, 17, 195–199. [Google Scholar] [CrossRef] [PubMed]
- PHAC. Fluwatch, Weekly Influenza reports: Fluwatch Summary, February 25, 2018 to March 3, 2018 (Week 9). Available online: https://www.canada.ca/en/public-health/services/diseases/flu-influenza/influenza-surveillance/weekly-influenza-reports.html (accessed on 14 March 2018).
- CDC. Fluview; Weekly U.S. Influenza Surveillance Report. Available online: https://www.cdc.gov/flu/weekly/ (accessed on December 2018).
- Most, J.; Weiss, G. Consecutive Infections With Influenza A and B Virus in Children During the 2014-2015 Seasonal Influenza Epidemic. J. Infect. Dis. 2016, 214, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, K.; Cieslak, K.; Kowalczyk, D.; Brydak, L.B. Co-infection with Influenza Viruses and Influenza-Like Virus During the 2015/2016 Epidemic Season. Adv. Exp. Med. Biol. 2017, 968, 7–12. [Google Scholar]
- WHO. Influenza Updates. Available online: http://www.who.int/influenza/surveillance_monitoring/updates/en/ (accessed on December 2018).
- PHAC. An Advisory Committee Statement (ACS) National Advisory Committee on Immunization (NACI): Canadian Immunization Guide Chapter on Influenza and Statement on Seasonal Influenza Vaccine for 2016–2017. Available online: https://www.canada.ca/content/dam/phac-aspc/migration/phac-aspc/naci-ccni/assets/pdf/flu-2016-2017-grippe-eng.pdf (accessed on December 2018).
- CDC. Preventing Seasonal Flu With Vaccination. Available online: https://www.cdc.gov/flu/protect/vaccine/ (accessed on December 2018).
- Cohen, J. Why is the flu vaccine so mediocre? Science 2017, 357, 1222–1223. [Google Scholar] [CrossRef]
- Hoskins, T.W.; Davies, J.R.; Smith, A.J.; Miller, C.L.; Allchin, A. Assessment of inactivated influenza-A vaccine after three outbreaks of influenza A at Christ’s Hospital. Lancet 1979, 1, 33–35. [Google Scholar] [CrossRef]
- Skowronski, D.M.; Chambers, C.; De Serres, G.; Sabaiduc, S.; Winter, A.L.; Dickinson, J.A.; Gubbay, J.B.; Drews, S.J.; Fonseca, K.; Charest, H.; et al. Vaccine effectiveness against lineage matched and mismatched influenza B viruses across 8 seasons in Canada, 2010–2011 to 2017–2018. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B.; Gruber, W.C.; Mendelman, P.M.; Mehta, H.B.; Mahmood, K.; Reisinger, K.; Treanor, J.; Zangwill, K.; Hayden, F.G.; Bernstein, D.I.; et al. Correlates of immune protection induced by live, attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine. J. Infect. Dis. 2000, 181, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Legge, A.; Dodds, L.; MacDonald, N.E.; Scott, J.; McNeil, S. Rates and determinants of seasonal influenza vaccination in pregnancy and association with neonatal outcomes. CMAJ 2014, 186, E157–E164. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Kwong, J.C.; Regan, A.K.; Katz, M.A.; Drews, S.J.; Azziz-Baumgartner, E.; Klein, N.P.; Chung, H.; Effler, P.V.; Feldman, B.S.; et al. Influenza Vaccine Effectiveness in Preventing Influenza-associated Hospitalizations During Pregnancy: A Multi-country Retrospective Test Negative Design Study, 2010–2016. Clin. Infec.t Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.L.; Mendelman, P.M.; Mallon, K.P.; Jackson, L.A.; Gorse, G.J.; Belshe, R.B.; Glezen, W.P.; Wittes, J. Effectiveness of live, attenuated intranasal influenza virus vaccine in healthy, working adults: A randomized controlled trial. JAMA 1999, 282, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yan, A.W.; Heffernan, J.M.; Petrie, S.; Moss, R.G.; Carolan, L.A.; Guarnaccia, T.A.; Kelso, A.; Barr, I.G.; McVernon, J.; et al. Innate Immunity and the Inter-exposure Interval Determine the Dynamics of Secondary Influenza Virus Infection and Explain Observed Viral Hierarchies. PLoS Comput. Biol. 2015, 11, e1004334. [Google Scholar] [CrossRef] [PubMed]
- WHO. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Zhang, X.; Deriaud, E.; Jiao, X.; Braun, D.; Leclerc, C.; Lo-Man, R. Type I interferons protect neonates from acute inflammation through interleukin 10-producing B cells. J. Exp. Med. 2007, 204, 1107–1118. [Google Scholar] [CrossRef]
- Sun, C.M.; Deriaud, E.; Leclerc, C.; Lo-Man, R. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity 2005, 22, 467–477. [Google Scholar] [CrossRef]
- Belderbos, M.E.; Levy, O.; Stalpers, F.; Kimpen, J.L.; Meyaard, L.; Bont, L. Neonatal plasma polarizes TLR4-mediated cytokine responses towards low IL-12p70 and high IL-10 production via distinct factors. PLoS ONE 2012, 7, e33419. [Google Scholar] [CrossRef]
- Agrons, G.A.; Courtney, S.E.; Stocker, J.T.; Markowitz, R.I. From the archives of the AFIP: Lung disease in premature neonates: Radiologic-pathologic correlation. Radiographics 2005, 25, 1047–1073. [Google Scholar] [CrossRef]
- Langley, J.M.; Frenette, L.; Chu, L.; McNeil, S.; Halperin, S.; Li, P.; Vaughn, D. A randomized, controlled non-inferiority trial comparing A(H1N1)pmd09 vaccine antigen, with and without AS03 adjuvant system, co-administered or sequentially administered with an inactivated trivalent seasonal influenza vaccine. BMC Infect. Dis. 2012, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Langley, J.M.; Wang, L.; Aggarwal, N.; Bueso, A.; Chandrasekaran, V.; Cousin, L.; Halperin, S.A.; Li, P.; Liu, A.; McNeil, S.; et al. Immunogenicity and Reactogenicity of an Inactivated Quadrivalent Influenza Vaccine Administered Intramuscularly to Children 6 to 35 Months of Age in 2012–2013: A Randomized, Double-Blind, Controlled, Multicenter, Multicountry, Clinical Trial. J. Pediatr. Infect. Dis. Soc. 2015, 4, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Collie, M.H.; Rushton, D.I.; Sweet, C.; Smith, H. Studies of influenza virus infection in newborn ferrets. J. Med. Microbiol. 1980, 13, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Paquette, S.G.; Banner, D.; Huang, S.S.; Almansa, R.; Leon, A.; Xu, L.; Bartoszko, J.; Kelvin, D.J.; Kelvin, A.A. Influenza Transmission in the Mother-Infant Dyad Leads to Severe Disease, Mammary Gland Infection, and Pathogenesis by Regulating Host Responses. PLoS Pathog. 2015, 11, e1005173. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Banner, D.; Degousee, N.; Leon, A.J.; Xu, L.; Paquette, S.G.; Kanagasabai, T.; Fang, Y.; Rubino, S.; Rubin, B.; et al. Differential pathological and immune responses in newly weaned ferrets are associated with a mild clinical outcome of pandemic 2009 H1N1 infection. J. Virol. 2012, 86, 13187–13201. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Moreno, J.; Carragher, D.M.; de la Luz Garcia-Hernandez, M.; Hwang, J.Y.; Kusser, K.; Hartson, L.; Kolls, J.K.; Khader, S.A.; Randall, T.D. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat. Immunol. 2011, 12, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Dunhill, M. Postnatal growth of the lung. Thorax 17 1962, 17, 329–333. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Turner, D.; Pham, Q.; Wherry, E.J.; Lefrancois, L.; Farber, D.L. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011, 187, 5510–5514. [Google Scholar] [CrossRef]
- Bergmann, B.; Grimsholm, O.; Thorarinsdottir, K.; Ren, W.; Jirholt, P.; Gjertsson, I.; Martensson, I.L. Memory B cells in mouse models. Scand. J. Immunol. 2013, 78, 149–156. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infection Relationship | Example Sequence | Example Strain 1 | Example Strain 2 |
|---|---|---|---|
| Monosubtypic Homologous | sH1N1 -> sH1N1 | A/Brisbane/59/2007 | A/Brisbane/59/2007 |
| Monosubtypic Heterologous | sH1N1 -> 2009 H1N1 | A/Brisbane/59/2007 | A/California/07/2009 |
| Heterosubtypic | sH1N1 -> H3N2 | A/FortMonmouth/1/1947 | A/HongKong/1/1968 |
| Heterotypic | sH1N1 -> B-V virus | A/Brisbane/59/2007 | B/Brisbane/60/2008 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, M.E.; King, M.L.; Kelvin, A.A. Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections. Viruses 2019, 11, 122. https://doi.org/10.3390/v11020122
Francis ME, King ML, Kelvin AA. Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections. Viruses. 2019; 11(2):122. https://doi.org/10.3390/v11020122
Chicago/Turabian StyleFrancis, Magen Ellen, Morgan Leslie King, and Alyson Ann Kelvin. 2019. "Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections" Viruses 11, no. 2: 122. https://doi.org/10.3390/v11020122
APA StyleFrancis, M. E., King, M. L., & Kelvin, A. A. (2019). Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections. Viruses, 11(2), 122. https://doi.org/10.3390/v11020122