Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Characteristics of VZV Latency
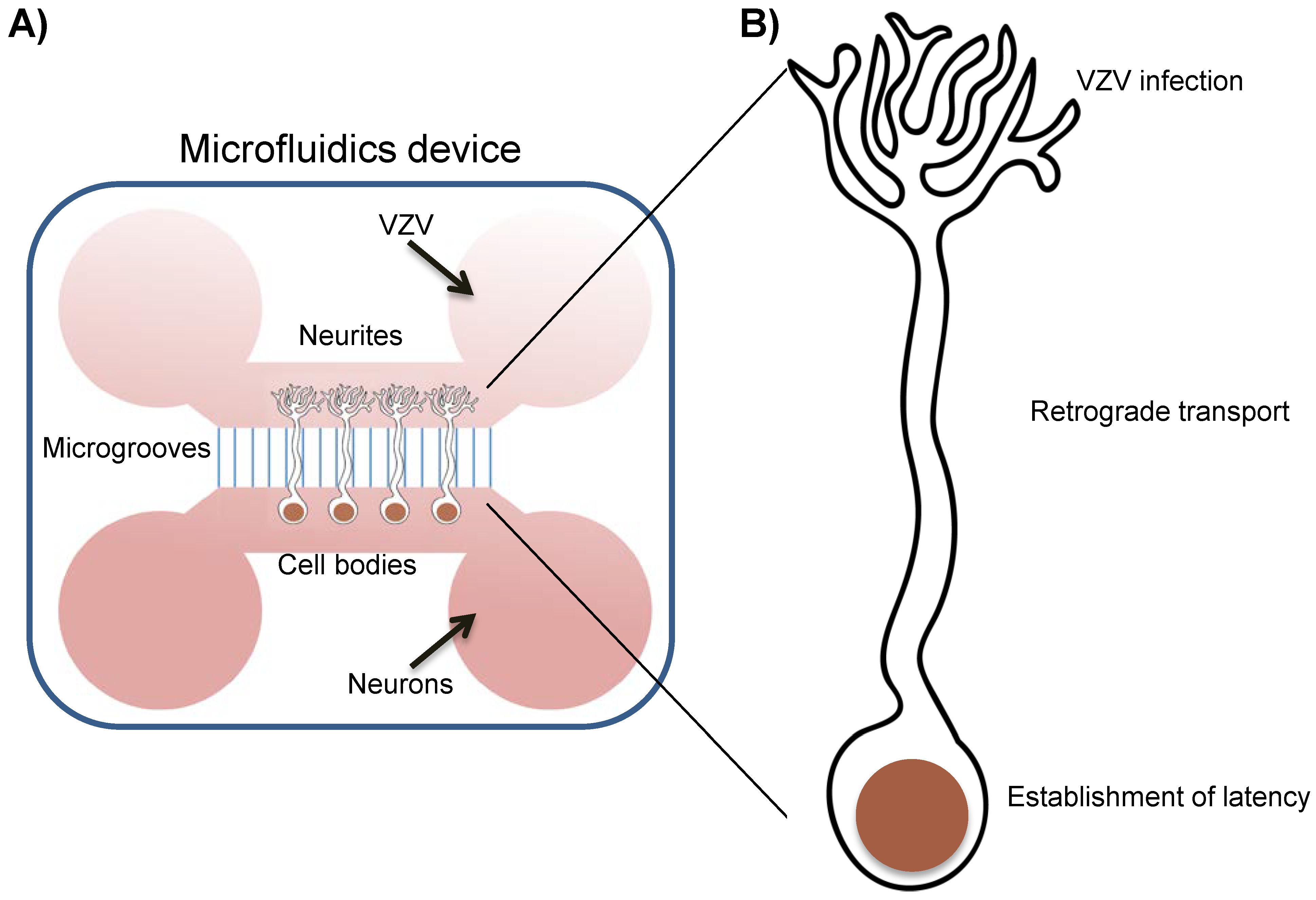
3. Generation and Characterization of Human Neurons to Study VZV
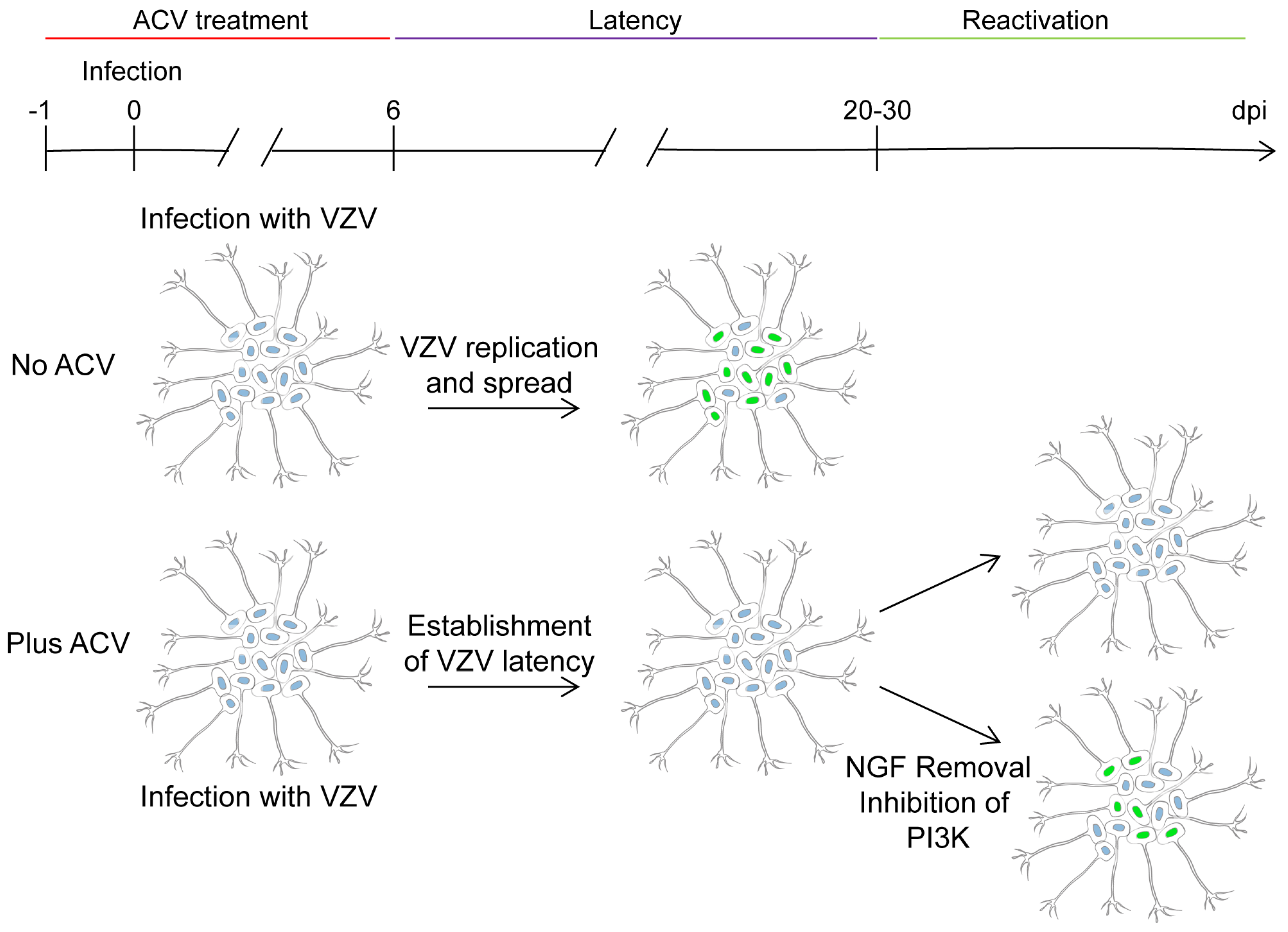
4. In vitro Latency/Reactivation Models
5. The Relevance of NGF in VZV Latency and Reactivation
6. Concluding Remarks and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Gershon, A.A.; Chen, J.; Davis, L.; Krinsky, C.; Cowles, R.; Reichard, R.; Gershon, M. Latency of varicella zoster virus in dorsal root, cranial, and enteric ganglia in vaccinated children. Trans. Am. Clin. Climatol. Assoc. 2012, 123, 17–33, discussion 33–35. [Google Scholar] [PubMed]
- Nagel, M.A.; Rempel, A.; Huntington, J.; Kim, F.; Choe, A.; Gilden, D. Frequency and abundance of alphaherpesvirus DNA in human thoracic sympathetic ganglia. J. Virol. 2014, 88, 8189–8192. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Chen, J.; Gershon, M.D. Use of saliva to identify varicella zoster virus infection of the gut. Clin. Infect. Dis. 2015, 61, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Gershon, A.A.; Li, Z.; Cowles, R.A.; Gershon, M.D. Varicella zoster virus (VZV) infects and establishes latency in enteric neurons. J. Neurovirol. 2011, 17, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Azarkh, Y.; Bos, N.; Gilden, D.; Cohrs, R.J. Human trigeminal ganglionic explants as a model to study alphaherpesvirus reactivation. J. Neurovirol. 2012, 18, 456–461. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Badani, H.; Bos, N.; Scianna, C.; Hoskins, I.; Baird, N.L.; Gilden, D. Alphaherpesvirus DNA replication in dissociated human trigeminal ganglia. J. Neurovirol. 2016, 22, 688–694. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Badani, H.; Baird, N.L.; White, T.M.; Sanford, B.; Gilden, D. Induction of varicella zoster virus DNA replication in dissociated human trigeminal ganglia. J. Neurovirol. 2017, 23, 152–157. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.; Choe, A.; Nagel, M.A.; Gilden, D.; Osterhaus, A.D.; Cohrs, R.J.; Verjans, G.M. Restricted varicella-zoster virus transcription in human trigeminal ganglia obtained soon after death. J. Virol. 2012, 86, 10203–10206. [Google Scholar] [CrossRef]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Depledge, D.P.; Sadaoka, T.; Ouwendijk, W.J.D. Molecular aspects of varicella-zoster virus latency. Viruses 2018, 10, 349. [Google Scholar] [CrossRef]
- Gilden, D.H.; Dueland, A.N.; Devlin, M.E.; Mahalingam, R.; Cohrs, R. Varicella-zoster virus reactivation without rash. J. Infect. Dis. 1992, 166 (Suppl. 1), S30–S34. [Google Scholar] [CrossRef]
- Kennedy, P.G.E.; Gershon, A.A. Clinical features of varicella-zoster virus infection. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Mehta, S.K.; Schmid, D.S.; Gilden, D.H.; Pierson, D.L. Asymptomatic reactivation and shed of infectious varicella zoster virus in astronauts. J. Med. Virol. 2008, 80, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.K.; Cohrs, R.J.; Forghani, B.; Zerbe, G.; Gilden, D.H.; Pierson, D.L. Stress-induced subclinical reactivation of varicella zoster virus in astronauts. J. Med. Virol. 2004, 72, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Gershon, M.D. Pathogenesis and current approaches to control of varicella-zoster virus infections. Clin. Microbiol. Rev. 2013, 26, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Sauerbrei, A. Diagnosis, antiviral therapy, and prophylaxis of varicella-zoster virus infections. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Sharp, M.; Maecker, H.T.; Maino, V.C.; Arvin, A.M. Frequencies of memory t cells specific for varicella-zoster virus, herpes simplex virus, and cytomegalovirus by intracellular detection of cytokine expression. J. Infect. Dis. 2000, 181, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Koenig, H.C.; Garland, J.M.; Weissman, D.; Mounzer, K. Vaccinating hiv patients: Focus on human papillomavirus and herpes zoster vaccines. AIDS Rev. 2013, 15, 77–86. [Google Scholar] [PubMed]
- Levin, M.J.; Smith, J.G.; Kaufhold, R.M.; Barber, D.; Hayward, A.R.; Chan, C.Y.; Chan, I.S.; Li, D.J.; Wang, W.; Keller, P.M.; et al. Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-dose vzv vaccine. J. Infect. Dis. 2003, 188, 1336–1344. [Google Scholar] [CrossRef]
- Saylor, D.; Thakur, K.; Venkatesan, A. Acute encephalitis in the immunocompromised individual. Curr. Opin. Infect. Dis. 2015, 28, 330–336. [Google Scholar] [CrossRef]
- Weinberg, A.; Levin, M.J. Vzv t cell-mediated immunity. Curr. Top. Microbiol. Immunol. 2010, 342, 341–357. [Google Scholar] [PubMed]
- Zhang, Y.; Cosyns, M.; Levin, M.J.; Hayward, A.R. Cytokine production in varicella zoster virus-stimulated limiting dilution lymphocyte cultures. Clin. Exp. Immunol. 1994, 98, 128–133. [Google Scholar] [CrossRef]
- Kristie, T.M.; Liang, Y.; Vogel, J.L. Control of alpha-herpesvirus ie gene expression by hcf-1 coupled chromatin modification activities. Biochim. Biophys. Acta 2010, 1799, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kristie, T.M.; Vogel, J.L.; Sears, A.E. Nuclear localization of the c1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl. Acad. Sci. USA 1999, 96, 1229–1233. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, W.; Lorentzen, E.U.; Eing, B.R.; Muller, M.; King, N.J.; Klupp, B.; Mettenleiter, T.C.; Kuhn, J.E. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 2012, 8, e1002679. [Google Scholar] [CrossRef]
- Gary, L.; Gilden, D.H.; Cohrs, R.J. Epigenetic regulation of varicella-zoster virus open reading frames 62 and 63 in latently infected human trigeminal ganglia. J. Virol. 2006, 80, 4921–4926. [Google Scholar] [CrossRef]
- Clarke, P.; Beer, T.; Cohrs, R.; Gilden, D.H. Configuration of latent varicella-zoster virus DNA. J. Virol. 1995, 69, 8151–8154. [Google Scholar] [PubMed]
- Nagel, M.A.; Choe, A.; Traktinskiy, I.; Cordery-Cotter, R.; Gilden, D.; Cohrs, R.J. Varicella-zoster virus transcriptome in latently infected human ganglia. J. Virol. 2011, 85, 2276–2287. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Grinfeld, E.; Bell, J.E. Varicella-zoster virus gene expression in latently infected and explanted human ganglia. J. Virol. 2000, 74, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Gilden, D.H.; Kinchington, P.R.; Grinfeld, E.; Kennedy, P.G. Varicella-zoster virus gene 66 transcription and translation in latently infected human ganglia. J. Virol. 2003, 77, 6660–6665. [Google Scholar] [CrossRef] [PubMed]
- Lungu, O.; Panagiotidis, C.A.; Annunziato, P.W.; Gershon, A.A.; Silverstein, S.J. Aberrant intracellular localization of varicella-zoster virus regulatory proteins during latency. Proc. Natl. Acad. Sci. USA 1998, 95, 7080–7085. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Wellish, M.; Cohrs, R.; Debrus, S.; Piette, J.; Rentier, B.; Gilden, D.H. Expression of protein encoded by varicella-zoster virus open reading frame 63 in latently infected human ganglionic neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Proenca, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sobel, R.A.; Lai, M.; Triglia, R.; Steain, M.; Abendroth, A.; Arvin, A. Apparent expression of varicella-zoster virus proteins in latency resulting from reactivity of murine and rabbit antibodies with human blood group a determinants in sensory neurons. J. Virol. 2012, 86, 578–583. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.; Flowerdew, S.E.; Wick, D.; Horn, A.K.; Sinicina, I.; Strupp, M.; Osterhaus, A.D.; Verjans, G.M.; Hufner, K. Immunohistochemical detection of intra-neuronal vzv proteins in snap-frozen human ganglia is confounded by antibodies directed against blood group a1-associated antigens. J. Neurovirol. 2012, 18, 172–180. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Gershon, M.D. A model of lytic, latent, and reactivating varicella-zoster virus infections in isolated enteric neurons. J. Infect. Dis. 2008, 197 (Suppl. 2), S61–S65. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Ranayhossaini, D.J.; Deluca, N.A. Activities of icp0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J. Virol. 2011, 85, 4993–5002. [Google Scholar] [CrossRef] [PubMed]
- Halford, W.P.; Kemp, C.D.; Isler, J.A.; Davido, D.J.; Schaffer, P.A. Icp0, icp4, or vp16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 2001, 75, 6143–6153. [Google Scholar] [CrossRef] [PubMed]
- Halford, W.P.; Schaffer, P.A. Icp0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 2001, 75, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein icp0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 1993, 67, 7501–7512. [Google Scholar]
- Preston, C.M. Reactivation of expression from quiescent herpes simplex virus type 1 genomes in the absence of immediate-early protein icp0. J. Virol. 2007, 81, 11781–11789. [Google Scholar] [CrossRef]
- Miller, C.S.; Danaher, R.J.; Jacob, R.J. Icp0 is not required for efficient stress-induced reactivation of herpes simplex virus type 1 from cultured quiescently infected neuronal cells. J. Virol. 2006, 80, 3360–3368. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sawtell, N.M. Evidence that the herpes simplex virus type 1 icp0 protein does not initiate reactivation from latency in vivo. J. Virol. 2006, 80, 10919–10930. [Google Scholar] [CrossRef]
- Christensen, J.; Steain, M.; Slobedman, B.; Abendroth, A. Differentiated neuroblastoma cells provide a highly efficient model for studies of productive varicella-zoster virus infection of neuronal cells. J. Virol. 2011, 85, 8436–8442. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Montague, P.; Scott, F.; Grinfeld, E.; Ashrafi, G.H.; Breuer, J.; Rowan, E.G. Varicella-zoster viruses associated with post-herpetic neuralgia induce sodium current density increases in the nd7-23 nav-1.8 neuroblastoma cell line. PLoS ONE 2013, 8, e51570. [Google Scholar] [CrossRef]
- Baird, N.L.; Yu, X.; Cohrs, R.J.; Gilden, D. Varicella zoster virus (VZV)-human neuron interaction. Viruses 2013, 5, 2106–2115. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, S.C. Neural subtype specification from human pluripotent stem cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef]
- Kawasaki, H.; Mizuseki, K.; Nishikawa, S.; Kaneko, S.; Kuwana, Y.; Nakanishi, S.; Nishikawa, S.I.; Sasai, Y. Induction of midbrain dopaminergic neurons from es cells by stromal cell-derived inducing activity. Neuron 2000, 28, 31–40. [Google Scholar] [CrossRef]
- Pomp, O.; Brokhman, I.; Ben-Dor, I.; Reubinoff, B.; Goldstein, R.S. Generation of peripheral sensory and sympathetic neurons and neural crest cells from human embryonic stem cells. Stem Cells 2005, 23, 923–930. [Google Scholar] [CrossRef]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brustle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human es and ips cells by dual inhibition of smad signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lonnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell rna sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Li, C.L.; Li, K.C.; Wu, D.; Chen, Y.; Luo, H.; Zhao, J.R.; Wang, S.S.; Sun, M.M.; Lu, Y.J.; Zhong, Y.Q.; et al. Somatosensory neuron types identified by high-coverage single-cell rna-sequencing and functional heterogeneity. Cell Res. 2016, 26, 967. [Google Scholar] [CrossRef]
- Zerboni, L.; Arvin, A. Neuronal subtype and satellite cell tropism are determinants of varicella-zoster virus virulence in human dorsal root ganglia xenografts in vivo. PLoS Pathog. 2015, 11, e1004989. [Google Scholar] [CrossRef]
- Yu, X.; Seitz, S.; Pointon, T.; Bowlin, J.L.; Cohrs, R.J.; Jonjic, S.; Haas, J.; Wellish, M.; Gilden, D. Varicella zoster virus infection of highly pure terminally differentiated human neurons. J. Neurovirol. 2013, 19, 75–81. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Nair, S.; Velmurugan, K.; Liang, Q.; Mahalingam, R.; Cohrs, R.J.; Nagel, M.A.; Gilden, D. Varicella-zoster virus infection of differentiated human neural stem cells. J. Virol. 2011, 85, 6678–6686. [Google Scholar] [CrossRef]
- Baird, N.L.; Bowlin, J.L.; Hotz, T.J.; Cohrs, R.J.; Gilden, D. Interferon gamma prolongs survival of varicella-zoster virus-infected human neurons in vitro. J. Virol. 2015, 89, 7425–7427. [Google Scholar] [CrossRef]
- Baird, N.L.; Bowlin, J.L.; Yu, X.; Jonjic, S.; Haas, J.; Cohrs, R.J.; Gilden, D. Varicella zoster virus DNA does not accumulate in infected human neurons. Virology 2014, 458–459, 1–3. [Google Scholar] [CrossRef]
- Goodwin, T.J.; McCarthy, M.; Osterrieder, N.; Cohrs, R.J.; Kaufer, B.B. Three-dimensional normal human neural progenitor tissue-like assemblies: A model of persistent varicella-zoster virus infection. PLoS Pathog. 2013, 9, e1003512. [Google Scholar] [CrossRef]
- Baird, N.L.; Bowlin, J.L.; Cohrs, R.J.; Gilden, D.; Jones, K.L. Comparison of varicella-zoster virus rna sequences in human neurons and fibroblasts. J. Virol. 2014, 88, 5877–5880. [Google Scholar] [CrossRef]
- Sloutskin, A.; Kinchington, P.R.; Goldstein, R.S. Productive vs non-productive infection by cell-free varicella zoster virus of human neurons derived from embryonic stem cells is dependent upon infectious viral dose. Virology 2013, 443, 285–293. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Slobedman, B.; Cunningham, A.L.; Miranda-Saksena, M.; Boadle, R.A.; Abendroth, A. Productive varicella-zoster virus infection of cultured intact human ganglia. J. Virol. 2007, 81, 6752–6756. [Google Scholar] [CrossRef]
- Markus, A.; Grigoryan, S.; Sloutskin, A.; Yee, M.B.; Zhu, H.; Yang, I.H.; Thakor, N.V.; Sarid, R.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: Direct demonstration of axonal infection, transport of vzv, and productive neuronal infection. J. Virol. 2011, 85, 6220–6233. [Google Scholar] [CrossRef]
- Lee, K.S.; Zhou, W.; Scott-McKean, J.J.; Emmerling, K.L.; Cai, G.Y.; Krah, D.L.; Costa, A.C.; Freed, C.R.; Levin, M.J. Human sensory neurons derived from induced pluripotent stem cells support varicella-zoster virus infection. PLoS ONE 2012, 7, e53010. [Google Scholar] [CrossRef]
- Harkness, J.M.; Kader, M.; DeLuca, N.A. Transcription of the herpes simplex virus 1 genome during productive and quiescent infection of neuronal and nonneuronal cells. J. Virol. 2014, 88, 6847–6861. [Google Scholar] [CrossRef]
- Wilcox, C.L.; Johnson, E.M., Jr. Characterization of nerve growth factor-dependent herpes simplex virus latency in neurons in vitro. J. Virol. 1988, 62, 393–399. [Google Scholar]
- Wilcox, C.L.; Johnson, E.M., Jr. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol. 1987, 61, 2311–2315. [Google Scholar]
- Cliffe, A.R.; Wilson, A.C. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J. Virol. 2017, 91, e01419-16. [Google Scholar] [CrossRef]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates hsv-1 latency in neurons. Cell Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient reversal of episome silencing precedes vp16-dependent transcription during reactivation of latent hsv-1 in neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An in vitro model of latency and reactivation of varicella zoster virus in human stem cell-derived neurons. PLoS Pathog. 2015, 11, e1004885. [Google Scholar] [CrossRef]
- Grose, C.; Brunel, P.A. Varicella-zoster virus: Isolation and propagation in human melanoma cells at 36 and 32 degrees c. Infect. Immun. 1978, 19, 199–203. [Google Scholar]
- Sadaoka, T.; Depledge, D.P.; Rajbhandari, L.; Venkatesan, A.; Breuer, J.; Cohen, J.I. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc. Natl. Acad. Sci. USA 2016, 113, E2403–E2412. [Google Scholar] [CrossRef]
- Kurapati, S.; Sadaoka, T.; Rajbhandari, L.; Jagdish, B.; Shukla, P.; Ali, M.A.; Kim, Y.J.; Lee, G.; Cohen, J.I.; Venkatesan, A. Role of the jnk pathway in varicella-zoster virus lytic infection and reactivation. J. Virol. 2017, 91, e00640-17. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; MacGibeny, M.A.; Hogue, I.B.; Enquist, L.W. Compartmented neuronal cultures reveal two distinct mechanisms for alpha herpesvirus escape from genome silencing. PLoS Pathog. 2017, 13, e1006608. [Google Scholar] [CrossRef]
- Bradshaw, R.A.; Pundavela, J.; Biarc, J.; Chalkley, R.J.; Burlingame, A.L.; Hondermarck, H. Ngf and prongf: Regulation of neuronal and neoplastic responses through receptor signaling. Adv. Biol. Regul. 2015, 58, 16–27. [Google Scholar] [CrossRef]
- Barbacid, M. Neurotrophic factors and their receptors. Curr. Opin. Cell Biol. 1995, 7, 148–155. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef]
- Yanez, A.A.; Harrell, T.; Sriranganathan, H.J.; Ives, A.M.; Bertke, A.S. Neurotrophic factors ngf, gdnf and ntn selectively modulate hsv1 and hsv2 lytic infection and reactivation in primary adult sensory and autonomic neurons. Pathogens 2017, 6. [Google Scholar] [CrossRef]
- St Leger, A.J.; Hendricks, R.L. Cd8+ t cells patrol hsv-1-infected trigeminal ganglia and prevent viral reactivation. J. Neurovirol. 2011, 17, 528–534. [Google Scholar] [CrossRef]
- De Regge, N.; Van Opdenbosch, N.; Nauwynck, H.J.; Efstathiou, S.; Favoreel, H.W. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Pourchet, A.; Modrek, A.S.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens 2017, 6, 24. [Google Scholar] [CrossRef]
- Como, C.N.; Pearce, C.M.; Cohrs, R.J.; Baird, N.L. Interleukin-6 and type 1 interferons inhibit varicella zoster virus replication in human neurons. Virology 2018, 522, 13–18. [Google Scholar] [CrossRef]
- Decman, V.; Kinchington, P.R.; Harvey, S.A.; Hendricks, R.L. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J. Virol. 2005, 79, 10339–10347. [Google Scholar] [CrossRef]
- Linderman, J.A.; Kobayashi, M.; Rayannavar, V.; Fak, J.J.; Darnell, R.B.; Chao, M.V.; Wilson, A.C.; Mohr, I. Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell Rep. 2017, 18, 1312–1323. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baird, N.L.; Zhu, S.; Pearce, C.M.; Viejo-Borbolla, A. Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation. Viruses 2019, 11, 103. https://doi.org/10.3390/v11020103
Baird NL, Zhu S, Pearce CM, Viejo-Borbolla A. Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation. Viruses. 2019; 11(2):103. https://doi.org/10.3390/v11020103
Chicago/Turabian StyleBaird, Nicholas L., Shuyong Zhu, Catherine M. Pearce, and Abel Viejo-Borbolla. 2019. "Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation" Viruses 11, no. 2: 103. https://doi.org/10.3390/v11020103
APA StyleBaird, N. L., Zhu, S., Pearce, C. M., & Viejo-Borbolla, A. (2019). Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation. Viruses, 11(2), 103. https://doi.org/10.3390/v11020103