Current In Vivo Models of Varicella-Zoster Virus Neurotropism

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Infection of Guinea Pigs with Varicella-Zoster Virus
2.1. Initial Approaches
2.2. First Guinea Pig Models of VZV
2.3. Recent Studies of VZV in Guinea Pigs
2.4. Concluding Remarks on the Guinea Pig Model
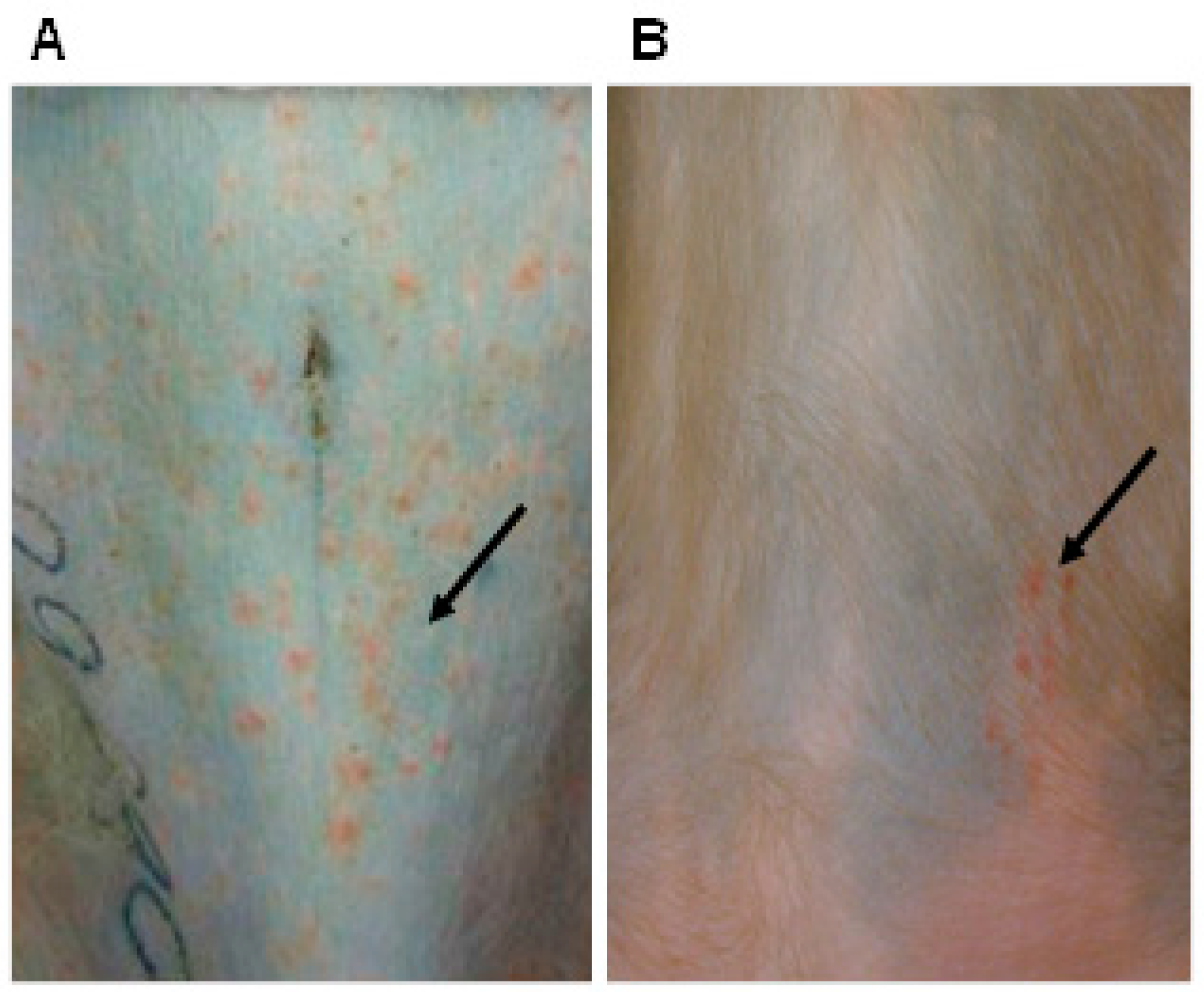
3. Cotton Rat Model
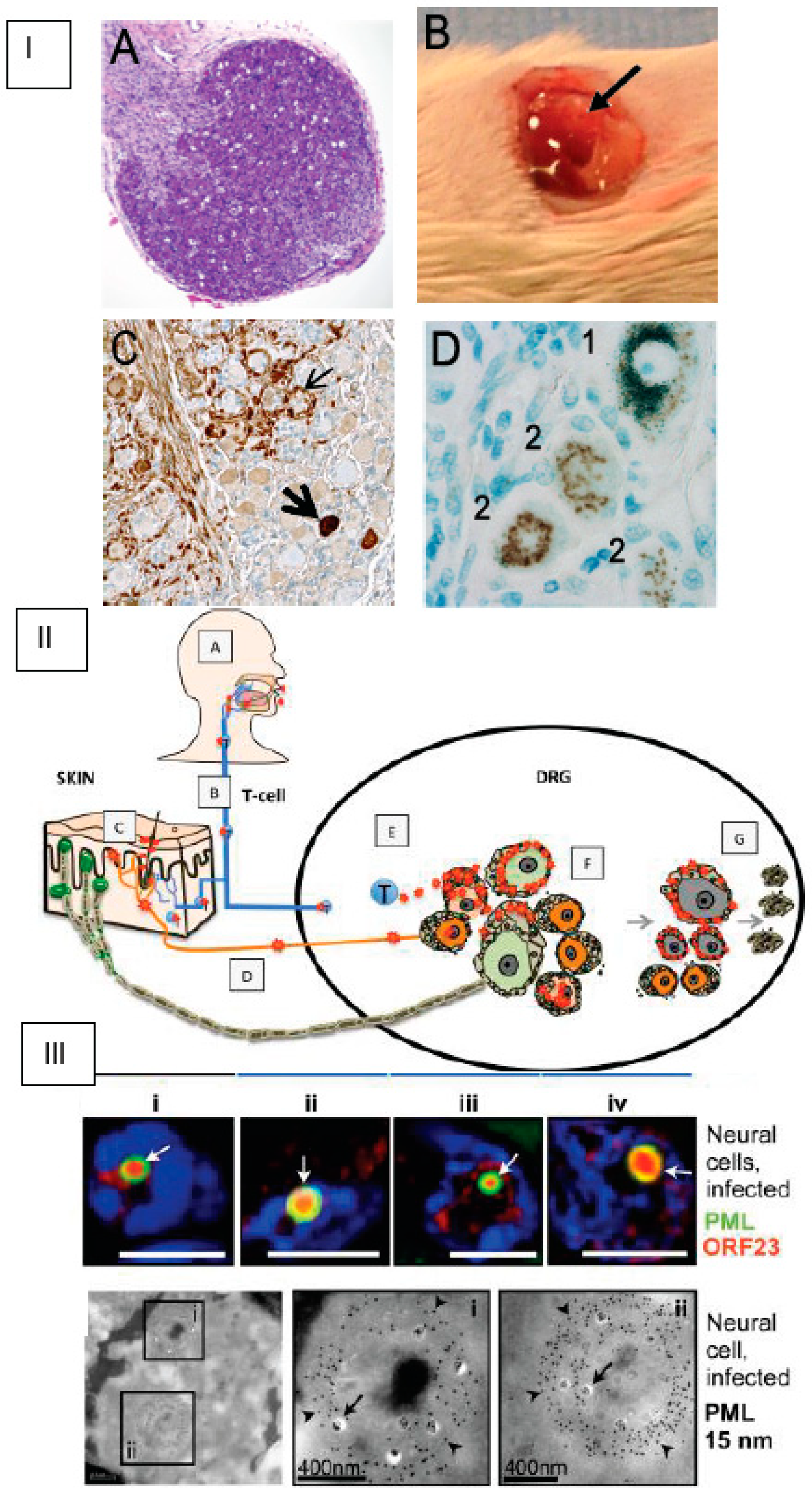
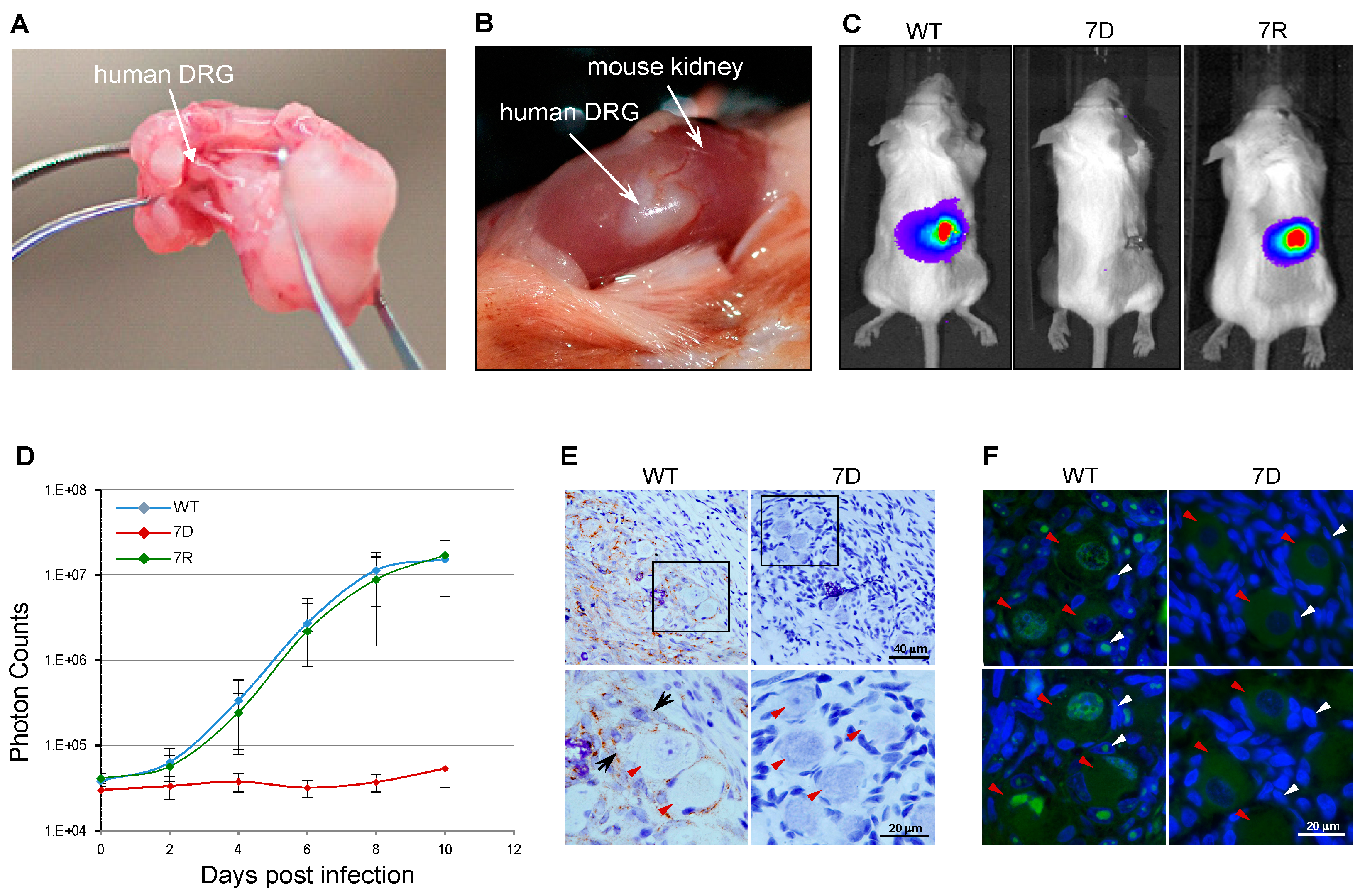
4. SCID-hu Mouse Model
Application of the SCID-hu Mouse Model to Study VZV ORF7 Neurotropism
5. Simian Varicella Virus (SVV) Infection in Non-Human Primates (NHP)
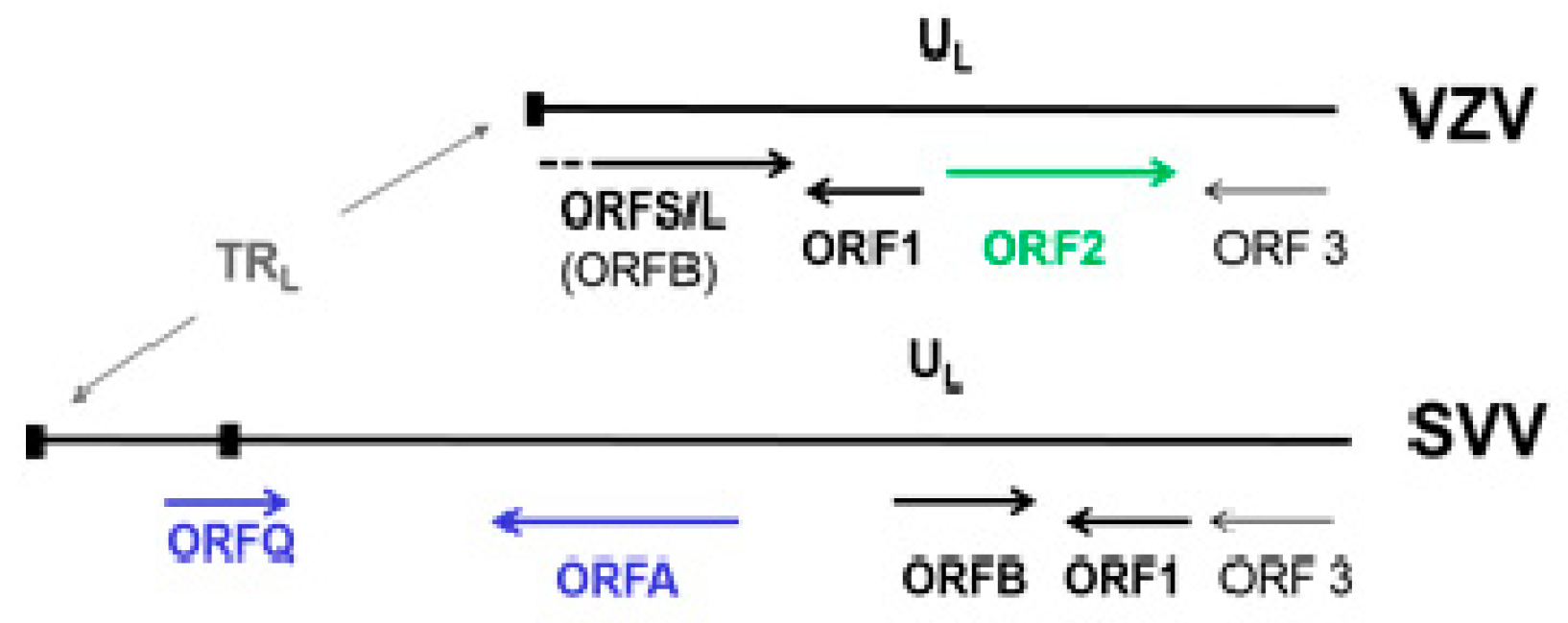
5.1. Molecular Aspects of SVV
5.2. Primary SVV Infection and Latency
5.3. SVV Reactivation in NHP
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takahashi, M.; Okuno, Y.; Otsuka, T.; Osame, J.; Takamizawa, A. Development of a live attenuated varicella vaccine. Biken J. 1975, 18, 25–33. [Google Scholar] [PubMed]
- Myers, M.; Duer, H.L.; Haulser, C.K. Experimental infection of guinea pigs with varicella-zoster virus. J. Infect. Dis. 1980, 142, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.; Stanberry, L.; Edmond, B. Varicella-zoster virus infection of strain 2 guinea pigs. J. Infect. Dis. 1985, 151, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Matsunga, Y.; Yamanishi, K.; Takahashi, M. Experimental infection and immune responses of guinea pigs with varicella zoster virus. Infect. Immun. 1982, 37, 407. [Google Scholar]
- Wroblewska, Z.; Devlin, M.; Reilly, K.; van Trieste, H.; Wellish, M.; Gilden, D.H. The production of varicella zoster virus antiserum in laboratory animals. Brief report. Arch. Virol. 1982, 74, 233–238. [Google Scholar] [CrossRef]
- Walz-Cicconi, M.A.; Rose, R.M.; Dammin, G.J.; Weller, T.H. Inoculation of guinea pigs with varicella-zoster virus via the respiratory route. Arch. Virol. 1986, 88, 265–277. [Google Scholar] [CrossRef]
- Weller, T.; Stoddard, M.B. Intranuclear inclusion bodies in cultures of human tissue inoculated with varicella vesicle fluid. J. Immunol. 1952, 68, 311–319. [Google Scholar] [PubMed]
- Fioretti, A.; Iwasaki, Y.; Furukawa, T.; Plotkin, S.A.; Kritchevsky, D. The growth of varicella-zoster virus in guinea pig embryo cells. Proc. Soc. Exp. Biol. Med. 1973, 144, 340–344. [Google Scholar] [CrossRef]
- Harbour, D.A.; Caunt, A.E. Infection of guinea-pig embryo cells with varicella-zoster virus. Arch. Virol. 1975, 49, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Otsuka, T.; Okuno, Y.; Asano, Y.; Yazaki, T.; Isomura, S. Live vaccine used to prevent the spread of varicella in children in hospital. Lancet 1974, 2, 1288–1290. [Google Scholar] [CrossRef]
- Gershon, A.A.; Steinberg, S.; Gelb, L.; Galasso, G.; Borkowsky, W.; LaRussa, P.; Farrara, A. Live attenuated varicella vaccine: efficacy for children with leukemia in remission. JAMA 1984, 252, 355–362. [Google Scholar] [CrossRef]
- Vazquez, M.; LaRussa, P.S.; Gershon, A.A.; Niccolai, L.M.; Muehlenbein, C.E.; Steinberg, S.P.; Shapiro, E.D. Effectiveness over time of varicella vaccine. JAMA 2004, 291, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.; Oxman, M.N.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Primers 2015, 1, 15016. [Google Scholar] [CrossRef]
- Oxman, M.N.; Levin, M.J.; Johnson, G.R.; Schmader, K.E.; Straus, S.E.; Gelb, L.D.; Arbeit, R.D.; Simberkoff, M.S.; Gershon, A.A.; Davis, L.E.; et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 2005, 352, 2271–2284. [Google Scholar] [CrossRef] [PubMed]
- Pavan-Langston, D.; Dunkel, E.C. Ocular varicella-zoster virus infection in the guinea pig. A new in vivo model. Arch. Ophthalmol. 1989, 107, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Wang, Y.; Nussenblatt, R.; Straus, S.E.; Hooks, J.J. Chronic uveitis in guinea pigs infected with varicella-zoster virus expressing E. coli β-galactosidase. J. Infect. Dis. 1998, 177, 293–300. [Google Scholar] [CrossRef]
- Myers, M.; Connelly, B.L. Animal models of varicella. J. Infect. Dis. 1992, 166, S48–S50. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Wang, Y.; Pesnicak, L.; Cohen, J.; Hooks, J.; Straus, S.; Williams, R.K. Recombinant VZV glycoproteins E and I: Immunologic responses and clearance of virus in a guinea pig model of chronic uveitis. J. Infect. Dis. 1998, 178, 310–317. [Google Scholar] [CrossRef]
- Myers, M.G.; Connelly, B.; Stanberry, L.R. Varicella in hairless guinea pigs. J. Infect. Dis. 1991, 163, 746–751. [Google Scholar] [CrossRef]
- Arvin, A.M.; Solem, S.; Koropchak, C.; Kinney-Thomas, E.; Paryani, S.G. Humoral and cellular immunity to varicella-zoster virus glycoprotein, gpI, and to a non-glycosylated protein p170, in strain 2 guinea pigs. J. Gen. Virol. 1987, 68, 2449–2454. [Google Scholar] [CrossRef]
- Hayward, A.; Burger, R.; Scheper, R.; Arvin, A. Major histocompatibility complex restriction of T-cell responses to varicella-zoster virus in guinea pigs. J. Virol. 1991, 65, 1491–1495. [Google Scholar]
- Lowry, P.W.; Solem, S.; Watson, B.N.; Koropchak, C.; Thackeray, H.; Kinchington, P.; Ruyechan, W.; Ling, P.; Hay, J.; Arvin, A. Immunity in strain 2 guinea pigs inoculated with vaccinia virus recombinants expressing varicella-zoster virus glycoproteins I, IV, V, or the protein product of the immediate early gene 62. J. Gen. Virol. 1992, 73, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Lowry, P.W.; Sabella, C.; Koropchak, C.; Watson, B.N.; Thackray, H.M.; Abruzzi, G.M.; Arvin, A.M. Investigation of the pathogenesis of varicella-zoster virus infection in guinea pigs by using polymerase chain reaction. J. Infect. Dis. 1993, 167, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Sabella, C.; Lowry, P.; Abbruzzi, G.; Koropchek, C.; Kinchington, P.; Sagedh-Zadeh, M.; Hay, J.; Ruyechan, W.; Arvin, A. Immunization with immediate-early tegument protein (open reading frame 62) of varicella-zoster virus protects guinea pigs against virus challenge. J. Virol. 1993, 67, 7673–7676. [Google Scholar] [PubMed]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Hope-Simpson, R.E. The nature of herpes zoster: a long term study and a new hypothesis. Proc. R. Soc. Med. 1965, 58, 9–20. [Google Scholar]
- Silverstein, S.; Straus, S.E. Pathogenesis of latency and reactivation. In Varicella-Zoster Virus: Virology and Clinical Management; Arvin, A., Gershon, A., Eds.; Cambridge University Press: Cambridge, UK, 2000; pp. 123–141. [Google Scholar]
- Kirchgessner, A.L.; Tamir, H.; Gershon, M.D. Identification and stimulation by serotonin of intrinsic sensory neurons of the submucosal plexus of the guinea pig gut: activity-induced expression of Fos immunoreactivity. J. Neurosci. 1992, 12, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kunze, W.A.; Bornstein, J.C.; Furness, J.B. Identification of sensory nerve cells in a peripheral organ (the intestine) of a mammal. Neuroscience 1995, 66, 1–4. [Google Scholar] [CrossRef]
- Chen, J.; Gershon, A.; Silverstein, S.J.; Li, Z.S.; Lungu, O.; Gershon, M.D. Latent and lytic infection of isolated guinea pig enteric and dorsal root ganglia by varicella zoster virus. J. Med. Virol. 2003, 70, S71–S78. [Google Scholar] [CrossRef]
- Gilden, D.H.; Gesser, R.; Smith, J.; Wellish, M.; Laguardia, J.J.; Cohrs, R.J.; Mahalingam, R. Presence of VZV and HSV-1 DNA in human nodose and celiac ganglia. Virus Genes 2001, 23, 145–147. [Google Scholar] [CrossRef]
- Richter, E.R.; Dias, J.K.; Gilbert, J.E., 2nd; Atherton, S.S. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J. Infect. Dis. 2009, 200, 1901–1906. [Google Scholar] [CrossRef]
- Chen, J.; Gershon, A.A.; Li, Z.; Cowles, R.A.; Gershon, M.D. Varicella zoster virus (VZV) infects and establishes latency in enteric neurons. J. Neurovirol. 2011, 17, 578–589. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Davis, L.; Krinsky, C.; Cowles, R.; Reichard, R.; Gershon, M.D. Latency of varicella zoster virus in dorsal root, cranial, and enteric ganglia in vaccinated children. Trans. Am. Clin. Climatol. Assoc. 2012, 123, 17–33. [Google Scholar]
- Gershon, A.A.; Chen, J.; Gershon, M.D. Use of saliva to identify varicella-zoster virus (VZV) infection of the gut. Clin. Infect. Dis. 2015, 61, 536–544. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Gershon, M.D. A model of lytic, latent, and reactivating varicella-zoster virus infections in isolated enteric neurons. J. Infect. Dis. 2008, 197 (Suppl. 2), S61–S65. [Google Scholar] [CrossRef]
- Gershon, A.A.; Gershon, M.D. The Jeremiah Metzger Lecture varicella zoster virus: from outside to inside. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 282–299. [Google Scholar]
- Mahalingam, R.; Messaoudi, I.; Gilden, D. Simian varicella virus pathogenesis. Curr. Top. Microbiol. Immunol. 2010, 342, 309–321. [Google Scholar]
- Gan, L.; Wang, M.; Chen, J.J.; Gershon, M.D.; Gershon, A.A. Infected peripheral blood mononuclear cells transmit latent varicella zoster virus infection to the guinea pig enteric nervous system. J. Neurovirol. 2014, 20, 442–456. [Google Scholar] [CrossRef]
- Gershon, M.; Gershon, A. Varicella-zoster virus and the enteric nervous system. J. Infect. Dis. 2018, 218 (Suppl. 2), S113–S119. [Google Scholar] [CrossRef]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.; Breuer, J.A. Spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Levin, M.J. Varicella-zoster virus and virus DNA in the blood and oropharynx of people with latent or active varicella-zoster virus infections. Jclin. Virol. 2014, 61, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.J.; Gilsdorf, J.R.; South, M.A.; Singleton, E.B. Gastritis as a complication of varicella. South. Med. J. 1973, 66, 539–541. [Google Scholar] [CrossRef]
- Morgan, E.R.; Smalley, L.A. Varicella in immunocompromised children: incidence of abdominal pain and organ involvement. Am. J. Dis. Child. 1983, 137, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Verdonck, L.F.; Cornelisssen, J.J.; Decker, A.W.; Rozenberg-Arska, M. Acute abdominal pain as a presenting symptom of varicella-zoster virus infection in recipients of bone marrow transplants. Clin. Infect. Dis. 1993, 16, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Magi, E. Severe varicella in an immunocompromised adult presenting with abdominal pain. West. J. Med. 2000, 173, 376–377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leena, M.; Ville, V.; Veli-Jukka, A. Visceral varicella zoster virus infection after stem cell transplantation: a possible cause of severe abdominal pain. Scand. J. Gastroenterol. 2006, 41, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Peritz, D.C.; Duncan, C.; Kurek, K.; Perez-Atayde, A.R.; Lehmann, L.E. Visceral varicella zoster virus (VZV) after allogeneic hematopoietic stem cell transplant (HSCT) in pediatric patients with chronic graft-versus-host disease (cGVHD). J. Pediatr. Hematol. Oncol. 2008, 30, 931–934. [Google Scholar] [CrossRef]
- Rau, R.; Fitzhugh, C.D.; Baird, K.; Cortez, K.J.; Li, L.; Fischer, S.H.; Cowen, E.W.; Balow, J.E.; Walsh, T.J.; Cohen, J.I.; et al. Triad of severe abdominal pain, inappropriate antidiuretic hormone secretion, and disseminated varicella-zoster virus infection preceding cutaneous manifestations after hematopoietic stem cell transplantation: utility of PCR for early recognition and therapy. Ped. Infect. Dis. J. 2008, 27, 265–268. [Google Scholar]
- Sato, H.; Pesnicak, L.; Cohen, J.I. Varicella-zoster virus ORF2 encodes a membrane phosphoprotein that is dispensable for viral replication and for establishment of latency. J. Virol. 2002, 76, 3575–3578. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Callanan, L.D.; Pesnicak, L.; Krogmann, T.; Cohen, J.I. Varicella-zoster virus (VZV) ORF17 protein induces RNA cleavage and is critical for replication of VZV at 37°C, but not 33°C. J. Virol. 2002, 76, 11012–11023. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Pesnicak, L.; Cohen, J.I. Use of a rodent model to show that varicella-zoster virus ORF61 is dispensable for establishment of latency. J. Med. Virol. 2003, 70, S79–S81. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Pesnicak, L.; Cohen, J.I. Varicella-zoster virus ORF47 protein kinase which is required for replication in human T cells, and ORF66 protein kinase which is expressed during latency, are dispensable for establishment of latency. J. Virol. 2003, 77, 11180–11185. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Srinivas, S.; Sato, H.; Pesnicak, L.; Straus, S.E.; Cohen, J.I. Varicella-zoster virus ORF21, which is expressed during latency, is essential for virus replication but dispensable for establishment of latency. J. Virol. 2003, 77, 1211–1218. [Google Scholar] [CrossRef]
- Cohen, J.I.; Cox, E.; Pesnicak, L.; Srinivas, S.; Krogmann, T. The varicella-zoster virus ORF63 latency-associated protein is critical for establishment of latency. J. Virol. 2004, 78, 11833–11840. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Krogmann, T.; Bontems, S.; Sadzot, C.; Pesnicak, L. Regions of the varicella-zoster virus ORF63 latency-associated protein important for efficient replication in vitro are also critical for efficient establishment of latency. J. Virol. 2005, 79, 5069–5077. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Krogmann, T.; Ross, J.P.; Pesnicak, L.P.; Prikhod’ko, E.A. The varicella-zoster virus ORF4 latency-associated protein is important for establishment of latency. J. Virol. 2005, 79, 6969–6975. [Google Scholar] [CrossRef]
- Cohen, J.I.; Krogmann, T.; Pesnicak, L.; Ali, M.A. Absence or overexpression of the varicella-zoster virus (VZV) ORF29 latency-associated protein impairs late gene expression and reduces latency in a rodent model. J. Virol. 2007, 81, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Gilden, D.H. Prevalence and abundance of latently transcribed varicella-zoster virus genes in human ganglia. J. Virol. 2007, 81, 2950–2956. [Google Scholar] [CrossRef]
- Ambagala, A.P.; Krogmann, T.; Qin, J.; Pesnicak, L.; Cohen, J.I. A varicella-zoster virus mutant impaired for latency in rodents, but not impaired for replication in cell culture. Virology 2010, 399, 194–200. [Google Scholar] [CrossRef][Green Version]
- Zerboni, L.; Ku, C.C.; Jones, C.D.; Zehnder, J.L.; Arvin, A.M. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6490–6495. [Google Scholar] [CrossRef]
- Zerboni, L.; Reichelt, M.; Jones, C.D.; Zehnder, J.L.; Ito, H.; Arvin, A.M. Aberrant infection and persistence of varicella-zoster virus in human dorsal root ganglia in vivo in the absence of glycoprotein I. Proc. Natl. Acad. Sci. USA 2007, 104, 14086–14091. [Google Scholar] [CrossRef]
- Zerboni, L.; Arvin, A. Investigation of varicella-zoster virus neurotropism and neurovirulence using SCID mouse-human DRG xenografts. J. Neurovirol. 2011, 17, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Arvin, A. Neuronal subtype and satellite cell tropism are determinants of varicella-zoster virus virulence in human dorsal root ganglia xenografts in vivo. Plos Pathog. 2015, 11, e1004989. [Google Scholar] [CrossRef]
- Zerboni, L.; Berarducci, B.; Rajamani, J.; Jones, C.D.; Zehnder, J.L.; Arvin, A. Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID mouse-human model of neuropathogenesis. J. Virol. 2011, 85, 98–111. [Google Scholar] [CrossRef]
- Reichelt, M.; Zerboni, L.; Arvin, A.M. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J. Virol. 2008, 82, 3971–3983. [Google Scholar] [CrossRef]
- Oliver, S.L.; Zerboni, L.; Sommer, M.; Rajamani, J.; Arvin, A.M. Development of recombinant varicella-zoster viruses expressing luciferase fusion proteins for live in vivo imaging in human skin and dorsal root ganglia xenografts. J. Virol. Methods 2008, 154, 182–193. [Google Scholar] [CrossRef][Green Version]
- Esiri, M.M.; Tomlinson, A.H. Herpes Zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J. Neuro Sci. 1972, 15, 35–48. [Google Scholar] [CrossRef]
- Zerboni, L.; Che, X.; Reichelt, M.; Qiao, Y.; Gu, H.; Arvin, A. Herpes simplex virus 1 tropism for human sensory ganglion neurons in the severe combined immunodeficiency mouse model of neuropathogenesis. J. Virol. 2013, 87, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Bertke, A.S.; Ma, A.; Margolis, M.S.; Margolis, T.P. Different mechanisms regulate productive herpes simplex virus 1 (HSV-1) and HSV-2 infections in adult trigeminal neurons. J. Virol. 2013, 87, 6512–6516. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.; Choe, A.; Nagel, M.A.; Gilden, D.; Osterhaus, A.D.; Cohrs, R.J.; Verjans, G.M. Restricted varicella-zoster virus transcription in human trigeminal ganglia obtained soon after death. J. Virol. 2012, 86, 10203–10206. [Google Scholar] [CrossRef]
- Zerboni, L.; Sobel, R.A.; Lai, M.; Triglia, R.; Steain, M.; Abendroth, A.; Arvin, A. Apparent expression of varicella-zoster virus proteins in latency resulting from reactivity of murine and rabbit antibodies with human blood group a determinants in sensory neurons. J. Virol. 2012, 86, 578–583. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.; Flowerdew, S.E.; Wick, D.; Horn, A.K.; Sinicina, I.; Strupp, M.; Osterhaus, A.D.; Verjans, G.M.; Hufner, K. Immunohistochemical detection of intra-neuronal VZV proteins in snap-frozen human ganglia is confounded by antibodies directed against blood group A1-associated antigens. J. Neurovirol. 2012, 18, 172–180. [Google Scholar] [CrossRef]
- Reichelt, M.; Wang, L.; Sommer, M.; Perrino, J.; Nour, A.M.; Sen, N.; Baiker, A.; Zerboni, L.; Arvin, A.M. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. Plos Pathog. 2011, 7, e1001266. [Google Scholar] [CrossRef]
- Cole, N.L.; Grose, C. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 2003, 13, 207–222. [Google Scholar] [CrossRef]
- Zhang, Z.; Rowe, J.; Wang, W.; Sommer, M.; Arvin, A.; Moffat, J.; Zhu, H. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J. Virol. 2007, 81, 9024–9033. [Google Scholar] [CrossRef]
- Zhang, Z.; Selariu, A.; Warden, C.; Huang, G.; Huang, Y.; Zaccheus, O.; Cheng, T.; Xia, N.; Zhu, H. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. Plos Pathog. 2010, 6, e1000971. [Google Scholar] [CrossRef] [PubMed]
- Selariu, A.; Cheng, T.; Tang, Q.; Silver, B.; Yang, L.; Liu, C.; Ye, X.; Markus, A.; Goldstein, R.; Cruz-Cosme, R.; et al. ORF7 of varicella-zoster virus Is a neurotropic factor. J. Virol. 2012, 86, 8614–8624. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.L. Simian varicella: a model for human varicella-zoster virus infections. Rev. Med. Virol. 2004, 14, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.L.; Gusick, N.J. Viral isolates derived from simian varicella epizootics are genetically related but distinct from other primate herpesviruses. Virology 1996, 224, 161–166. [Google Scholar] [CrossRef][Green Version]
- Schmidt, N.J. Improved yields and assay of simian varicella virus, and a comparison of certain biological properties of simian and human varicella viruses. J. Virol. Meth. 1982, 5, 229–241. [Google Scholar] [CrossRef]
- Weller, T.H. Serial propagation in vitro of agents producing inclusion bodies derived from varicella and herpes zoster. Proc. Soc. Exp. Biol. Med. 1953, 83, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, T.M., III; Gray, W.L. Simian varicella virus: Characterization of virion and infected cell polypeptides and the antigenic cross-reactivity with varicella-zoster virus. J. Gen. Virol. 1992, 73, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.J.; Arvin, A.M.; Martin, D.P.; Gard, E.P. Serological investigation of an outbreak of simian varicella in Erythrocebus patas monkeys. J. Clin. Microbiol. 1983, 18, 901–904. [Google Scholar] [PubMed]
- Felsenfeld, A.D.; Schmidt, N.J. Varicella-zoster virus immunizes patas monkeys against simian varicella-like disease. J. Gen. Virol. 1979, 42, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.L.; Oakes, J.E. Simian varicella virus DNA shares homology with human varicella-zoster virus DNA. Virology 1984, 136, 241–246. [Google Scholar] [CrossRef]
- Gray, W.L.; Pumphrey, C.Y.; Ruyechan, W.T.; Fletcher, T.M. The simian varicella virus and varicella zoster virus genomes are similar in size and structure. Virology 1992, 186, 562–572. [Google Scholar] [CrossRef]
- Pumphrey, C.Y.; Gray, W.L. The genomes of simian varicella virus and varicella zoster virus are colinear. Virus Res. 1992, 26, 255–266. [Google Scholar] [CrossRef]
- Gray, W.L.; Mullis, L.B.; Soike, K.F. Expression of the simian varicella virus glycoprotein E. Virus Res. 2001, 79, 27–37. [Google Scholar] [CrossRef]
- Gray, W.L.; Starnes, B.; White, M.W.; Mahalingam, R. The DNA sequence of the simian varicella virus genome. Virology 2001, 284, 123–130. [Google Scholar] [CrossRef]
- Mahalingam, R.; Gray, W.L. The simian varicella virus genome contains an invertible 665 base pair terminal element that is absent in the varicella zoster virus genome. Virology 2007, 366, 387–393. [Google Scholar] [CrossRef][Green Version]
- Ou, Y.; Gray, W.L. Simian varicella virus gene 28 and 29 promoters share a common upstream stimulatory factor-binding site and are induced by IE62 transactivation. J. Gen. Virol. 2006, 87, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.L.; Davis, K.; Ou, Y.; Ashburn, C.; Ward, T.M. Simian varicella virus gene 61 encodes a viral transactivator but is non-essential for in vitro replication. Arch. Virol. 2007, 152, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Pumphrey, C.Y.; Gray, W.L. Identification and analysis of the simian varicella virus thymidine kinase gene. Arch. Virol. 1996, 141, 43–55. [Google Scholar] [CrossRef]
- Ashburn, C.V.; Gray, W.L. Identification and characterization of the simian varicella virus uracil DNA glycosylase. Arch. Virol. 1999, 144, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, C.V.; Gray, W.L. Expression of the simian varicella virus glycoprotein L and H. Arch. Virol. 2001, 147, 335–348. [Google Scholar] [CrossRef]
- Gray, W.L.; Byrne, B.H. Characterization of the simian varicella virus glycoprotein C, which is nonessential for in vitro replication. Arch. Virol. 2003, 148, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Pumphrey, C.Y.; Gray, W.L. DNA sequence and transcriptional analysis of the simian varicella virus glycoprotein B gene. J. Gen. Virol. 1994, 75, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Kerns, A.; Barrin, A.; Kreklywich, C.; Streblow, D.N.; Messaoudi, I. Simian varicella virus gene expression during acute and latent infection of RM. J. Neurovirol. 2011, 17, 600–612. [Google Scholar] [CrossRef]
- Ou, Y.; Davis, K.A.; Traina-Dorge, V.; Gray, W.L. Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 2007, 81, 8149–8156. [Google Scholar] [CrossRef]
- Gray, W.L.; Mahalingam, R. A cosmid-based system for inserting mutations and foreign genes into the simian varicella virus genome. J. Virol. Meth. 2005, 130, 89–94. [Google Scholar] [CrossRef]
- Ward, T.M.; Williams, M.V.; Traina-Dorge, V.; Gray, W.L. The simian varicella virus uracil DNA glycosylase and dUTPase genes are expressed in vivo, but are non-essential for replication in cell culture. Virus Res. 2009, 142, 78–84. [Google Scholar] [CrossRef][Green Version]
- Ou, Y.; Traina-Dorge, V.; Davis, K.A.; Gray, W.L. Recombinant simian varicella vaccines induce immune responses to simian immunodeficiency virus (SIV) antigens in immunized vervet monkeys. Virology 2007, 364, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.L.; Zhous, F.; Noffke, J.; Tischer, B.K. Cloning the simian varicella virus genome in E. coli as an infectious bacterial artificial chromosome. Arch. Virol. 2011, 156, 739–746. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brazeau, E.; Wellish, M.; Kaufer, B.B.; Tischer, B.K.; Gray, W.; Zhou, F.; Osterrieder, N.; Hanlon, T.; Golive, A.; Hall, T.; et al. Simian varicella virus open reading frame 63/70 expression is required for efficient virus replication in culture. J. Neurovirol. 2011, 17, 274–280. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahalingam, R.; Kaufer, B.B.; Ouwendijk, W.J.D.; Verjans, G.M.G.M.; Coleman, C.; Hunter, M.; Palmer, B.E.; Clambey, E.; Nagel, M.A.; Traina-Dorge, V. Attenuation of simian varicella virus infection by enhanced green fluorescent protein in RM. J. Virol. 2018, 92, e02253-17. [Google Scholar] [CrossRef]
- Meyer, C.; Dewane, J.; Haberthur, K.; Engelmann, F.; Arnold, N.; Gray, W.; Messaoudi, I. Bacterial artificial chromosome derived simian varicella virus is pathogenic in vivo. Virol. J. 2013, 10, 278. [Google Scholar] [CrossRef]
- Kolappaswamy, K.; Mahalingam, R.; Traina-Dorge, V.; Shipley, S.T.; Gilden, D.H.; Kleinschmidt-Demasters, B.K.; McCleod, C.G., Jr.; Hungerford, L.L.; DeTolla, L.J. Disseminated simian varicella virus infection in an irradiated rhesus macaque (Macaca mulatta). J. Virol. 2007, 81, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Haberthur, K.; Messaoudi, I. Animal models of varicella zoster virus infection. Pathogens 2013, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Traina-Dorge, V.; Wellish, M.; Smith, J.; Gilden, D.H. Naturally acquired simian varicella virus infection in African green monkeys. J. Virol. 2002, 76, 8548–8550. [Google Scholar] [CrossRef]
- White, T.M.; Mahalingam, R.; Traina-Dorge, V.; Gilden, D.H. Simian varicella virus DNA is present and transcribed months after experimental infection of adult African green monkeys. J. Neurovirol. 2002, 8, 191–203. [Google Scholar] [CrossRef]
- Gray, W.L. Pathogenesis of simian varicella virus. J. Med. Virol. 2003, 70, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Barron, A.; Wellish, M.; Engelmann, F.; Legasse, A.; Planer, S.; Gilden, D.; Nikolich-Zugich, J.; Mahalingam, R. Simian varicella virus infection of RM recapitulates essential features of varicella zoster virus infection in humans. Plos Pathog. 2009, 5, e1000657. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Mahalingam, R.; de Swart, R.L.; Haagmans, B.L.; Van Amerongen, G.; Setu, S.; Gilden, D.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. T-cell tropism of simian varicella virus during primary infection. Plos Pathog. 2013, 9, e1003368. [Google Scholar] [CrossRef] [PubMed]
- Soike, K.F. Simian varicella virus infection in African and Asian monkeys. The potential for development of antivirals for animal diseases. Ann. N.Y. Acad. Sci. 1992, 653, 323–333. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Mahalingam, R.; Traina-Dorge, V.; van Amerongen, G.; Wellish, M.; Osterhaus, A.D.M.E.; Gilden, D.H.; Verjans, G.M.G.M. Simian varicella virus infection of Chinese RM produces ganglionic infection in the absence of rash. J. Neurovirol. 2012, 18, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.; Veazey, R.S.; Luckay, A.; Penedo, C.; Xu, K.; Lifson, J.D.; Marx, P.A. SIV(mac) pathogenesis in RM of Chinese and Indian origin compared with primary HIV infections in humans. AIDS 2002, 16, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Estep, R.D.; Messaoudi, I.; Wong, S.W. Simian herpesviruses and their risk to humans. Vaccine 2010, 28 (Suppl. 2), B78–B84. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gray, W.L.; Mullis, L.; Soike, K.F. Viral gene expression during acute simian varicella virus infection. J. Gen. Virol. 2002, 83, 841–846. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Traina-Dorge, V.; Doyle-Meyers, L.A.; Sanford, R.; Manfredo, J.; Blackmon, A.; Wellish, M.; James, S.; Alvarez, X.; Midkiff, C.; Palmer, B.E.; et al. Simian varicella virus is present in macrophages, dendritic cells and T cells in lymph nodes of RM after experimental reactivation. J. Virol. 2015, 89, 9817–9824. [Google Scholar] [CrossRef]
- Mahalingam, R.; Traina-Dorge, V.; Wellish, M.; Deharo, E.; Golive, A.; Messaoudi, I.; Gilden, D. Effect of time delay after necropsy on analysis of simian varicella virus expression in latently infected ganglia of RM. J. Virol. 2010, 84, 12454–12457. [Google Scholar] [CrossRef] [PubMed]
- Arnold, N.; Girke, T.; Sureshchandra, S.; Messaoudi, I. Acute simian varicella virus causes robust and sustained changes in gene expression in the sensory ganglia. J. Virol. 2016, 90, 10823–10843. [Google Scholar] [CrossRef] [PubMed]
- Haberthur, K.; Meyer, C.; Arnold, N.; Engelmann, F.; Jeske, D.R.; Messaoudi, I. Intrabronchial Infection of RM with simian varicella virus results in a robust immune response in the lungs. J. Virol. 2014, 88, 12777–12792. [Google Scholar] [CrossRef] [PubMed]
- Traina-Dorge, V.; Sanford, R.; James, S.; Doyle-Meyers, L.A.; de Haro, E.; Wellish, M.; Gilden, D.; Mahalingam, R. Robust pro-inflammatory and lesser anti-inflammatory immune responses during primary simian varicella virus infection and reactivation in RM. J. Neurovirol. 2014, 20, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Arnold, N.; Girke, T.; Sureshchandra, S.; Nguyen, C.; Rais, M.; Messaoudi, I. Genomic and functional analysis of the host response to acute simian varicella infection in the lung. Sci. Rep. 2016, 6, 34164. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.; Verjans, G.M. Pathogenesis of varicelloviruses in primates. J. Pathol. 2015, 235, 298–311. [Google Scholar] [CrossRef]
- Mahalingam, R.; Traina-Dorge, V.; Wellish, M.; Deharo, E.; Singletary, M.L.; Ribka, E.P.; Sanford, R.; Gilden, D. Latent simian varicella virus reactivates in monkeys treated with tacrolimus with or without exposure to irradiation. J. Neurovirol. 2010, 16, 342–354. [Google Scholar] [CrossRef] [PubMed]
- James, S.F.; Traina-Dorge, V.; Deharo, E.; Wellish, M.; Palmer, B.E.; Gilden, D.; Mahalingam, R. T cells increase before zoster and PD-1 expression increases at the time of zoster in immunosuppressed nonhuman primates latently infected with simian varicella virus. J. Neurovirol. 2014, 20, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Haberthur, K.; Kraft, A.; Arnold, N.; Park, B.; Meyer, C.; Aquith, M.; Dewane, J.; Messaoudi, I. Genome wide analysis of T cell responses during acute and latent simian varicella virus infections in RM. J. Virol. 2013, 87, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Haberthur, K.; Engelmann, F.; Park, B.; Barron, A.; Legasse, A.; Dewane, J.; Fischer, M.; Kerns, A.; Brown, M.; Messaoudi, I. CD4 T cell immunity is critical for the control of simian varicella virus infection in a nonhuman primate model of VZV infection. Plos Pathogens. 2011, 7, e1002367. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Dewane, J.; Kerns, A.; Haberthur, K.; Barron, A.; Park, B.; Messaoudi, I. Age and immune status of RM impact simian varicella virus gene expression in sensory ganglia. J. Virol. 2013, 87, 8294–8306. [Google Scholar] [CrossRef] [PubMed]
- Traina-Dorge, V.; Palmer, B.E.; Coleman, C.; Hunter, M.; Frieman, A.; Gilmore, A.; Altrock, K.; Doyle-Meyers, L.; Nagel, M.A.; Mahalingam, R. Reactivation of simian varicella virus in RM after CD4 T cell depletion. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Arnold, N.; Meyer, C.; Engelmann, F.; Messaoudi, I. Robust gene expression changes in the ganglia following subclinical reactivation in RM infected with simian varicella virus. J. Neurovirol. 2017, 23, 520–538. [Google Scholar] [CrossRef]
- Gray, W.L.; Williams, R.J.; Chang, R.; Soike, K.F. Experimental simian varicella virus infection of St. Kitts vervet monkeys. J. Med. Primatol. 1998, 27, 177–183. [Google Scholar] [CrossRef]
- Dueland, A.N.; Martin, J.R.; Devlin, M.E.; Wellish, M.; Mahalingam, R.; Cohrs, R.; Soike, K.F.; Gilden, D.H. Acute simian varicella virus infection: clinical, laboratory, pathologic, and virologic features. Lab. Invest. 1992, 66, 762–773. [Google Scholar]
- Kennedy, P.G.; Grinfeld, E.; Traina-Dorge, V.; Gilden, D.H.; Mahalingam, R. Neuronal localization of simian varicella virus DNA in ganglia of naturally infected African green monkeys. Virus Genes 2004, 28, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; van Veen, S.; Mehraban, T.; Mahalingam, R.; Verjans, G.M. Simian varicella virus infects enteric neurons and a4B7 Integrin expressing gut-tropic T-cells in nonhuman primates. Viruses 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Kerns, A.; Haberthur, K.; Dewane, J.; Walker, J.; Gray, W.; Messaoudi, I. Attenuation of adaptive immune response in rhesus macaques infected with simian varicella virus lacking open reading frame 61. J. Virol. 2013, 87, 2151–2163. [Google Scholar] [CrossRef] [PubMed]
- White, T.M.; Mahalingam, R.; Traina-Dorge, V.; Gilden, D.H. Persistence of simian varicella virus DNA in CD4+ and CD8+ blood mononuclear cells for years after intratracheal inoculation of African green monkeys. Virology 2002, 303, 192–198. [Google Scholar] [CrossRef]
- Arnold, N.; Messaoudi, I. Simian varicella virus causes robust transcriptional changes in T cells that support viral replication. Virus Res. 2017, 238, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Terada, K.; Kawano, S.; Yoshihiro, K.; Morita, T. Varicella-zoster virus (VZV) reactivation is related to the low response of VZV-specific immunity after chickenpox in infancy. Infect. Dis. 1994, 169, 650–652. [Google Scholar] [CrossRef]
- Rodriguez-Moreno, A.; Sanchez-Fructuoso, A.I.; Calvo, N.; Ridao, N.; Conesa, J.; Marques, M.; Prats, D.; Barrientos, A. Varicella infection in adult renal allograft recipients: experience at one center. Transplant. Proc. 2006, 38, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Habuka, M.; Wada, Y.; Kurosawa, Y.; Yamamoto, S.; Tani, Y.; Ohashi, R.; Ajioka, Y.; Nakano, M.; Narita, I. Fatal visceral disseminated varicella zoster infection during initial remission induction therapy in a patient with lupus nephritis and rheumatoid arthritis—possible association with mycophenolate mofetil and high-dose glucocorticoid therapy: a case report. Bmc Res. Notes 2018, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Fort, M.K.; Zeng, J.; Feily, A.; Ramirez-Pacheco, L.A.; Jenrette, J.M.; Mayhew, D.L.; Syed, T.; Cooper, S.L.; Linden, C.; Graybill, W.S.; et al. Radiotherapy-induced reactivation of neurotrophic human herpes viruses: Overview and management. J. Clin. Virol. 2018, 98, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, R.R.; Gillen, M.; Grant, R.; Liggitt, H.D.; Kiem, H.-P.; Kelley, S.T. Simian varicella virus in pigtailed macaques (Macaca nemestrina): Clinical, pathologic and virologic features. Comp. Med. 2009, 59, 482–487. [Google Scholar] [PubMed]
- Treuting, P.M.; Johnson-Delaney, C.; Birkebak, T.A. Diagnostic exercise: Vesicular epidermal rash, mucosal ulcerations and hepatic necrosis in a Cynomolgus monkey (Macaca Fasccicularis). Lab. Anim. Sci. 1998, 48, 384–386. [Google Scholar] [PubMed]
- Schoeb, T.T.; Eberle, R.; Black, D.H.; Parker, R.F.; Cartner, S.C. Diagnostic exercise: Papulovesicular dermatitis in RM (Macaca mulatta). Vet. Pathol. 2008, 45, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Traina-Dorge, V.; Wellish, M.; Lorino, R.; Sanford, R.; Ribka, E.P.; Alleman, S.J.; Brazeau, E.; Gilden, D.H. Simian varicella virus reactivation in Cynomolgus monkeys. Virology 2007, 368, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Ouwendkjk, W.J.; Abendroth, A.; Traina-Dorge, V.; Getu, S.; Steain, M.; Wellish, M.; Andeweg, A.C.; Osterhaus, A.D.M.E.; Gilden, D.; Verjans, G.M.G.M.; et al. T-cell infiltration correlates with CXCL10 expression in ganglia of cynomolgus macaques with reactivated simian varicella virus. J. Virol. 2013, 87, 2979–2982. [Google Scholar] [CrossRef] [PubMed]
- Steain, M.; Gowrishankar, K.; Rodiguez, M.; Slobedman, B.; Abendroth, A. Upregulation of CXCL10 in human dorsal root ganglia during experimental and natural varicella zoster virus infection. J. Virol. 2011, 85, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Zangeneh, Z.; Golmoghaddam, H.; Emad, M.; Erfani, N.; Doroudchi, M. Elevated PD-1 expression and decreased telomerase activity in memory T cells of patients with symptomatic herpes zoster infection. Cell. Mol. Biol. 2014, 60, 13–21. [Google Scholar]
- Zak-Prelich, M.; McKenzie, R.C.; Susa-Jedrzejowska, A.; Norval, M. Local immune responses and systemic cytokine responses in zoster: relationship to the development of postherpetic neurolgia. Ciln. Exp. Immunol. 2003, 131, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Traina-Dorge, V.; Mehta, S.; Rooney, B.; Crucian, B.; Doyle-Meyers, L.; Das, A.; Coleman, C.; Mahalingam, R. Simian varicella virus DNA in saliva and buccal cells after experimental infection rhesus macaques. Frontiers in Microb. 2019, (in press). [CrossRef]
- Guedon, J.-M.G.; Yee, M.B.; Zhang, M.; Harvey, S.A.K.; Goins, W.F.; Kinchington, P.R. Neuronal changes induced by varicella zoster virus in a rat model of postherpetic neuralgia. Virology 2015, 482, 167–180. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | Advantages | Disadvantages |
|---|---|---|
| Guinea pig | VZV infects guinea pig cells and can proliferate. VZV infects guinea pig enteric neurons in vitro and can establish latency if the inoculum contains a low multiplicity-of-infection in the absence of non-neuronal cells. VZV can be reactivated from latency in guinea pig enteric neurons in vitro by expressing ORF61. Intravenous inoculation of VZV-infected guinea pig or human lymphocytes infects guinea pigs and establishes latency in most, if not all, enteric and dorsal root ganglion neurons; animals remain symptom-free for months. A combination of immunosuppression with tacrolimus and a simulation of stress with intravenous corticotrophin-releasing hormone can reactivate VZV from latency. Guinea pigs are less expensive than NHP. Ganglia can be removed immediately after euthanasia, to avoid concerns about postmortem reactivation. | Guinea pigs do not display typical varicella. Reactivation elicits a severe disseminated syndrome, like disseminated zoster in humans, necessitating that the animals be euthanized. Guinea pigs have a long gestation period and thus are difficult and expensive to breed, complicating studies of congenital VZV and VZV in newborn animals. |
| Cotton rat | VZV replicates in cotton rat fibroblasts in vitro. VZV mutants can be tested. | Host proteins may not be conserved between cotton rats and humans, such that VZV proteins may not interact with cotton rat proteins the same way. |
| Animals are aggressive. | ||
| Limited species-specific immunologic reagents are available. | ||
| SCID-hu mouse | Differentiated human neurons and satellite cells, along with supportive tissues within the tissue microenvironment of the sensory ganglion, can be maintained long-term in vivo. Typical human neuronal subtypes are maintained, allowing analysis of VZV neurotropism at the single-cell level and comparison with HSV-1, the other neurotropic human alpha-herpesviruses. Viral determinants of VZV neurovirulence can be defined by evaluating VZV recombinants with targeted mutations to disrupt domains of VZV proteins. Opportunity to document the progression of VZV infection in sensory ganglia, demonstrating the capacity of VZV to induce fusion of neurons and satellite cells, unlike HSV-1. Opportunity to define innate defenses mounted against VZV infection by neural cells in vivo. Xenografts allow study of VZV infection of human DRG. Only available in vivo model for some human viruses. Better at reproducing the pathophysiology of human diseases and a better model for studying tissue tropism and antivirals. Relatively inexpensive compared to NHP. Allow monitoring and measuring viral replication in vivo using a bioluminescence assay. | Anterograde and retrograde transport of VZV between skin and ganglia cannot be modeled. Lacking human immune responses. Complicated surgery procedure. Large experimental variation. Human fetal tissues may be difficult to obtain. Absence of an adaptive immune system. |
| NHP (SVV) | Simian counterpart virus, SVV infects NHP, causes varicella, latent infection in ganglia and zoster that are similar to human VZV infection. | Expensive and need special approval. SVV reactivation results in a whole-body rash, while VZV reactivation in humans produces a dermatomal rash. SVV genome contains a unique ~3 kbp sequence at the left end, not present in VZV and potentially encoding species-specificity. Not the human virus, VZV. Pattern of infection is species-specific. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahalingam, R.; Gershon, A.; Gershon, M.; Cohen, J.I.; Arvin, A.; Zerboni, L.; Zhu, H.; Gray, W.; Messaoudi, I.; Traina-Dorge, V. Current In Vivo Models of Varicella-Zoster Virus Neurotropism. Viruses 2019, 11, 502. https://doi.org/10.3390/v11060502
Mahalingam R, Gershon A, Gershon M, Cohen JI, Arvin A, Zerboni L, Zhu H, Gray W, Messaoudi I, Traina-Dorge V. Current In Vivo Models of Varicella-Zoster Virus Neurotropism. Viruses. 2019; 11(6):502. https://doi.org/10.3390/v11060502
Chicago/Turabian StyleMahalingam, Ravi, Anne Gershon, Michael Gershon, Jeffrey I. Cohen, Ann Arvin, Leigh Zerboni, Hua Zhu, Wayne Gray, Ilhem Messaoudi, and Vicki Traina-Dorge. 2019. "Current In Vivo Models of Varicella-Zoster Virus Neurotropism" Viruses 11, no. 6: 502. https://doi.org/10.3390/v11060502
APA StyleMahalingam, R., Gershon, A., Gershon, M., Cohen, J. I., Arvin, A., Zerboni, L., Zhu, H., Gray, W., Messaoudi, I., & Traina-Dorge, V. (2019). Current In Vivo Models of Varicella-Zoster Virus Neurotropism. Viruses, 11(6), 502. https://doi.org/10.3390/v11060502