1. Introduction
Seoul virus (SEOV), which belongs to the genus
Orthohantavirus, family
Hantaviridae, and order
Bunyavirales, is enveloped, single-stranded, and a negative-sense RNA virus. The virus establishes a persistent infection, which causes no apparent harm in rodents [
1,
2]. However, SEOV is pathogenic, which caused hemorrhagic fever with renal syndrome (HFRS) in humans [
3,
4]. As the major endemic countries of HFRS, China accounts for 90% of total HFRS cases worldwide [
4,
5,
6,
7]. HFRS mainly occurred in rural areas in the past, but recently extended to urban areas and even to city centers in China [
4,
8,
9]. Unlike other hantaviruses, SEOV has a worldwide geographic distribution range, from Asia to Africa, Europe, and both Americas mainly because of worldwide spread of brown rats [
10]. Under the fast socioeconomic development and frequent trade, SEOV is probably the most important global hantavirus, which posed threats to public health in most areas [
10,
11]. However, SEOV remains the globally most underestimated pathogenic hantavirus [
11].
The SEOV genome consists of three separate segments, which is referred to as the large (L), medium (M), and small (S) segments. These three segments encode the viral RNA-dependent RNA polymerase (RdRP), two envelope glycoproteins (Gn and Gc), and nucleocapsid protein (N), respectively [
12,
13]. Generally, RNA viruses evolve with remarkable rapidity, high rates of mutation and substitution, short generation time, and huge population sizes [
14,
15]. Further evidences also show that purifying selection is a dominant evolutionary force for some RNA viral genes [
14,
15]. Additionally, recombination and reassortment might also play an important role in RNA virus evolution [
16,
17,
18]. Despite of some reports on hantaviruses prevalence and evolution [
19,
20,
21,
22], the sequence information and mechanisms of evolution in SEOV, especially in urban context, remain to be fully elucidated.
Urbanization is a global trend that results from economic development [
9,
23]. Features of urban environments provide unique opportunities for the rats to colonize, for example, suitable harborage, food and water sources, and efficient traffic systems [
24]. Additionally, close contacts occur between commensal rats and people in the urban environment, which further increases the risk of disease transmission [
9,
24,
25,
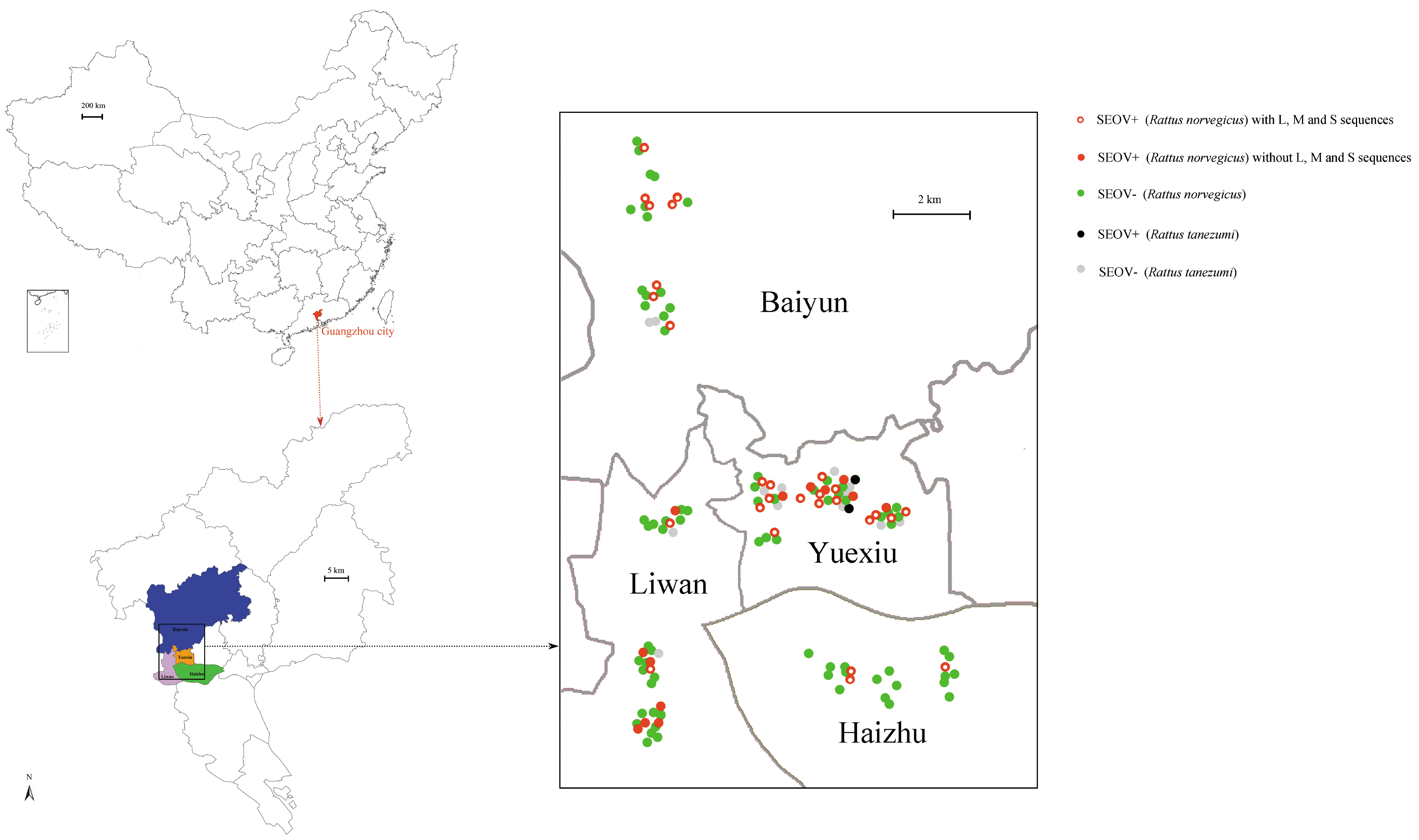
26]. Guangzhou has a population of 14.5 millionpeople, of which 20%–30% are floating (such as the temporary resident population, registered passenger population, and transit population). Indeed, the epidemic tendency of HFRS recently increased in Guangzhou [
27,
28]. For the first time we attempted to analyze the longer sequences of SEOV in the urban environment. By using different methods, we can gain more insights into the evolution of SEOV, which will provide effective guidance for relevant government departments.
4. Discussion
Numerous HFRS outbreaks are major environmental and public health concerns in the world, especially in developing countries [
9,
11,
23]. Although efforts have been made, the paucity of SEOV sequence data has hampered important researches. Here we implement an in-depth analysis of the genetic diversity and molecular evolution of SEOV in the urban environments.
The epidemiology investigation revealed that SEOV was the main hantavirusin Guangzhou city. This result was in agreement with former reports, which indicated that SEOV was predominant in the residential habitats in China [
4,
10,
11]. The amount of captured
R. norvegicus was higher than
R. tanezumi, suggesting that
R. norvegicus was the dominant species in Guangzhou city. However, the prevalence did not significantly differ between these two species. Our results confirmed that
R. norvegicus was the predominant carriers for hantaviruses in the residential habitats [
10,
11]. The high prevalence (33.33%) of SEOV may be directly related to the reservoir species and urban environment [
10]. Due to a combination of adequate harborage, food, and water supply, the population of city rats was high [
23,
24,
25]. Moreover, the frequent contact and dispersal of city rats might increase the spread of the virus, as we did not find any significant associations between geographical distance and genetic divergence in Guangzhou SEOV. Generally, weight is a good proxy for age [
49,
50]. The weights of positive ratswere significantly heavier than those of negative ones, which demonstrated that the older the animal, the greater the likelihood that it will become exposed to and infected with SEOV [
51]. Moreover, body weight has been used as an indicator of sexual maturity in
R. norvegicus. It meant that physiological and behavioral changes associated with sexual maturation may favor SEOV infection [
51].Unfortunately, we failed to recover the L, M, and S segments from
R. tanezumi and the other PCR-positive
R. norvegicus probably due to the sample quality. The rats in our study were trapped through snap traps and live traps. Thus, both dead and live rats were captured. We put snap traps the night before, checked and collected animals early in the next morning. Thus, the rats might be caught early and were dead for a long time, which caused the virus to degrade, further interfered with the amplification of gene segments. SEOV was a RNA virus, and thus the amplification of genome segments required high quality of tissues. Report shows that hantavirus is sensitive to heat (30 min at 60 °C) [
52] and the longevity of Puumala virus (PUUV) and Tula virus (TULV) depends on the temperature and moisture [
53]. In nature, there are additional physical and chemical factors, such as UV light, sunlight, and pH, which may influence the longevity of viruses [
53]. Moreover, no significant divergence was found between these successful or failed amplified SEOV on the basis of the part L segment. Particularly, we also recovered part of L, M, and S segments in other positive rats. The sequences were short and thus we did not use them for evolutionary analysis.
R. norvegicus and
R. tanezumi are partially sympatric in southern China [
54]. However, over the past 20 years,
R. tanezumi has significantly expanded northward in China and partially replaced the native
R. norvegicu [
54]. It will be a good example to clarify the adaptive evolution strategy of SEOV in southern and northern China, thus advancing the research of this severe human pathogen.
In our study, the best-fitting time-reversible model of nucleotide substitution was used for each gene segment to minimize the impact of site saturation. However, we still detected mutational saturation in the S segment of SEOV. Former studies showed that high levels of saturation were attained extremely rapidly in RNA virus [
35,
36]. Mutational saturation in the S segment may lead to incorrect estimation of evolutionary rates and time-scales in our study. For example, Saxenhofer et al. [
37] observed strong signals of mutational saturation at different levels of genetic divergence and they demonstrated that mutational saturation was the predominant cause of biases in molecular clock dating in PUUV and TULV [
37]. The mean rate of evolutionary change in SEOV was 10
−4 substitutions/site/year, which was similar to the rate of other RNA viruses [
15,
19,
21,
55]. For instance, the median substitution rate of PUUV was estimated as 2.70 × 10
−4 substitutions/site/year [
21]. Such a high substitution rate may be associated with the fact that SEOV relies on RNA-dependent RNA polymerase for replication, and lacks proofreading or base-excision repair mechanism [
14,
15]. Generally, the substitution rate is best described as the long-term rate at which genetic variants become fixed over evolutionary timescales. Hence, this rate reflects the complex interplay of natural selection, genetic drift, modes of transmission, and epidemiological processes [
14,
15], although sometimes displays a time-dependent bias in rate estimates [
35,
36,
37]. Conclusively, these observations suggest that urban SEOV is evolving substantially as fast as most RNA viruses.
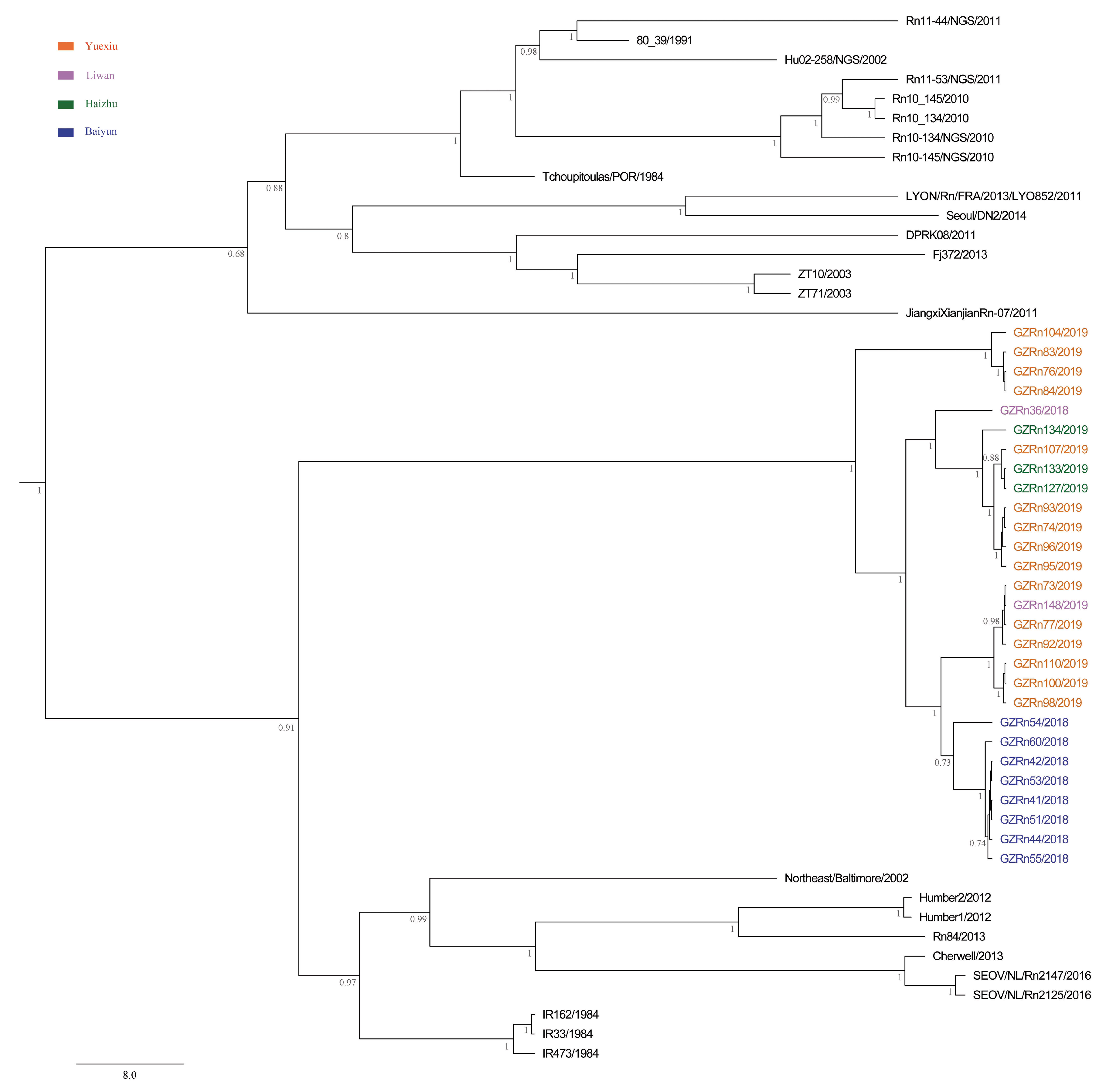
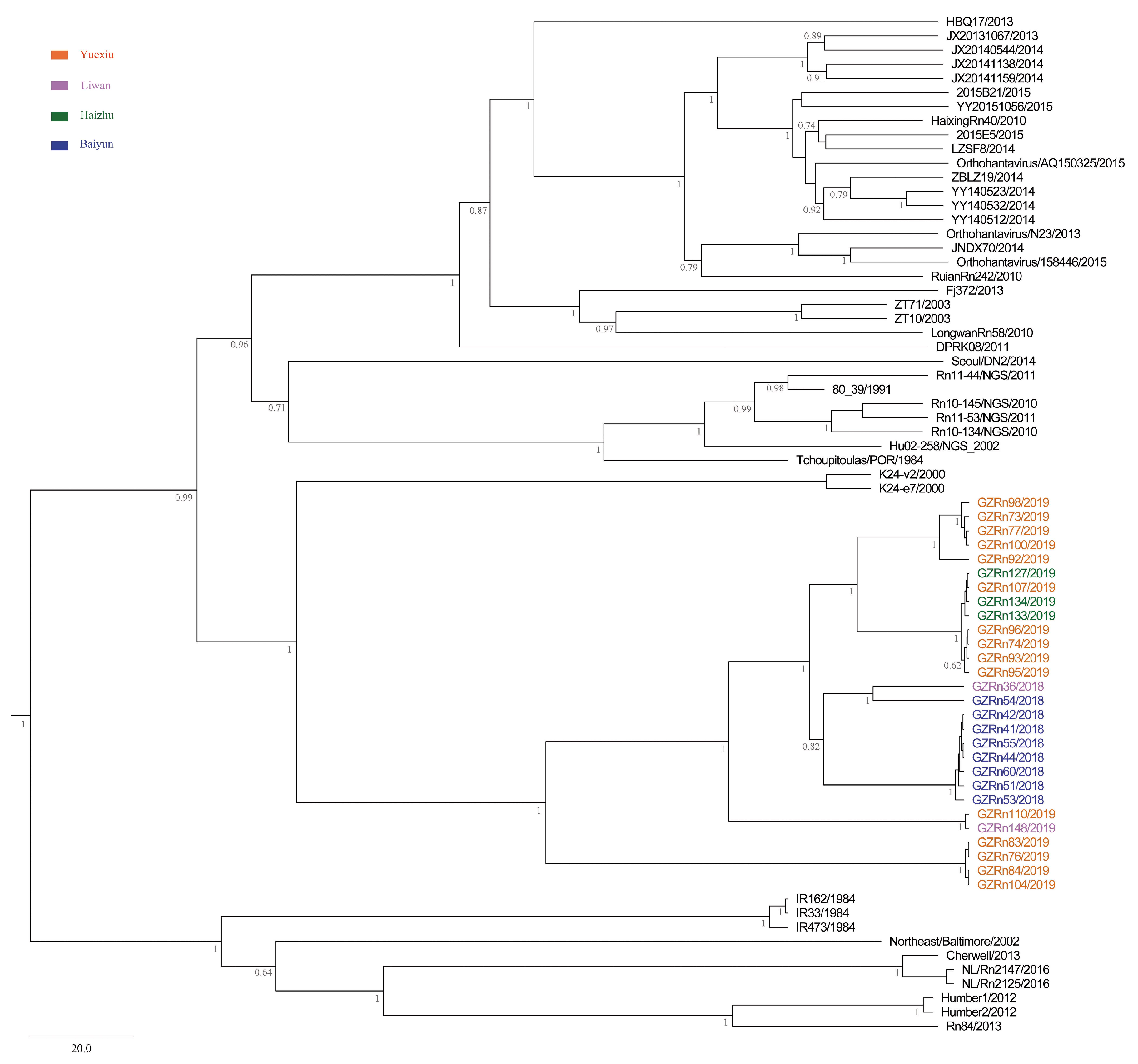
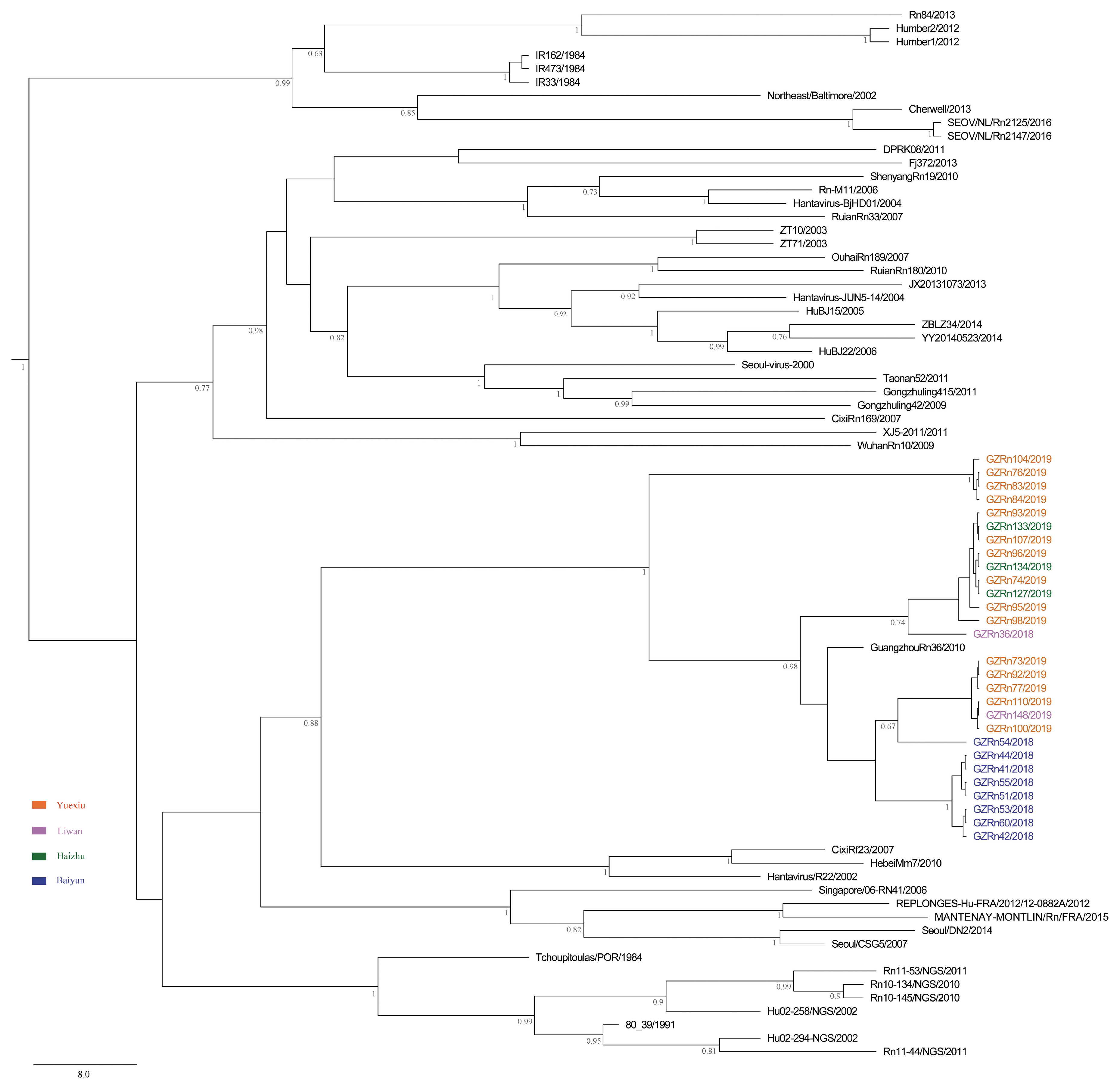
A better understanding of mechanisms that drive viral genetic variation may help us to understand the epidemiology [
14,
15,
19]. The molecular evolution of SEOV has long been overlooked. We found a high level of nucleotide polymorphisms in Guangzhou SEOV. The ratios of nonsynonymous to synonymous substitutions were very low for L, M, and S segments, indicating that most amino acid changes were deleterious, which would be removed by purifying selection. Thus, amino acid residues are preserved, while allowing near-neutral nucleotide sequences to continue evolving. Previous studies also revealed negative selection in other viruses [
56,
57,
58]. The positive selected codon 66 in the L segment may play an important role among these SEOV recovered from Guangzhou. For TULV in a natural hybrid zone, Saxenhofer et al. (2019) pointed to the positive selected codon 17 in the M segment [
59]. These positive selected sites may provide potential basis for the virus evolution. Phylogenetic analyses showed that Guangzhou SEOV clustered together in a single well-supported clade, indicating the genetic specificity of SEOV in Guangzhou. Environmental heterogeneity may shape the evolution of SEOV in Guangzhou. For example, phylogenetic analyses of PUUV typically detect genetic divergence even at relatively small geographical distances [
21]. This maybe because RNA viruses exert high evolutionary rates and genetic variability, and those variants which are adapted to different localities will be selected and fixed [
21,
60].
For many RNA viruses, recombination has been an important feature of their evolution [
16,
17]. In the present study, the genetic recombination was observed in L and M segments, and the former was first detected in nature. Moreover, GZRn95 and GZRn104 in L segment from the same sampling area had the same breakpoint, although their haplotypes were different. Particularly, GZRn98 was actually a double recombinant, because recombination was detected in L and M segments. These interesting findings are worth studying in the future. Mostowy et al. [
61] reported that the “recent recombinations” are determined as those that are present in some subset of strains in a lineage, while the “ancestral recombinations” are determined as those that are present between lineages that affect all strains in the lineage [
61]. We detected recombinations only in a few isolates of the Guangzhou strain. Thus, we inferred that these supposed events were recent. In addition, we detected no recombination in the S segment, which differed from another study conducted in Beijing [
5]. This might be attributed to the short sequence length of S segments in our study. Classically, viruses that generate persistent rather than acute infections may have high rate of recombination [
17]. Each hantavirus species associates closely with rodent species, where the virus establishes a persistent but asymptomatic infection [
1,
13,
62]. Therefore, recombination appears to be a common occurrence in hantaviruses [
17]. Recombination could lead to evolutionary changes [
16,
17]. For example, Xiao et al. [
63] demonstrated that recombination accelerated poliovirus adaptation and evolution, and was essential to enrich the population in beneficial mutations and to purge it from deleterious mutations. The recombination of L and M segments might be beneficial for SEOV fitness in Guangzhou, yet, remains to be studied.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}