EV71 Infection Induces IFNβ Expression in Neural Cells

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Viral Infection
2.3. Reagents
2.4. Immunofluorescence Staining
2.5. Protein Isolation and Western Blot
2.6. RNA Isolation and RT-PCR
2.7. siRNA Knockdown
2.8. In Vitro Proteinase Cleavage Assay
2.9. IFN-β Antibody Blocking Assay
2.10. Plaque Assay
2.11. Statistical Analysis
3. Results
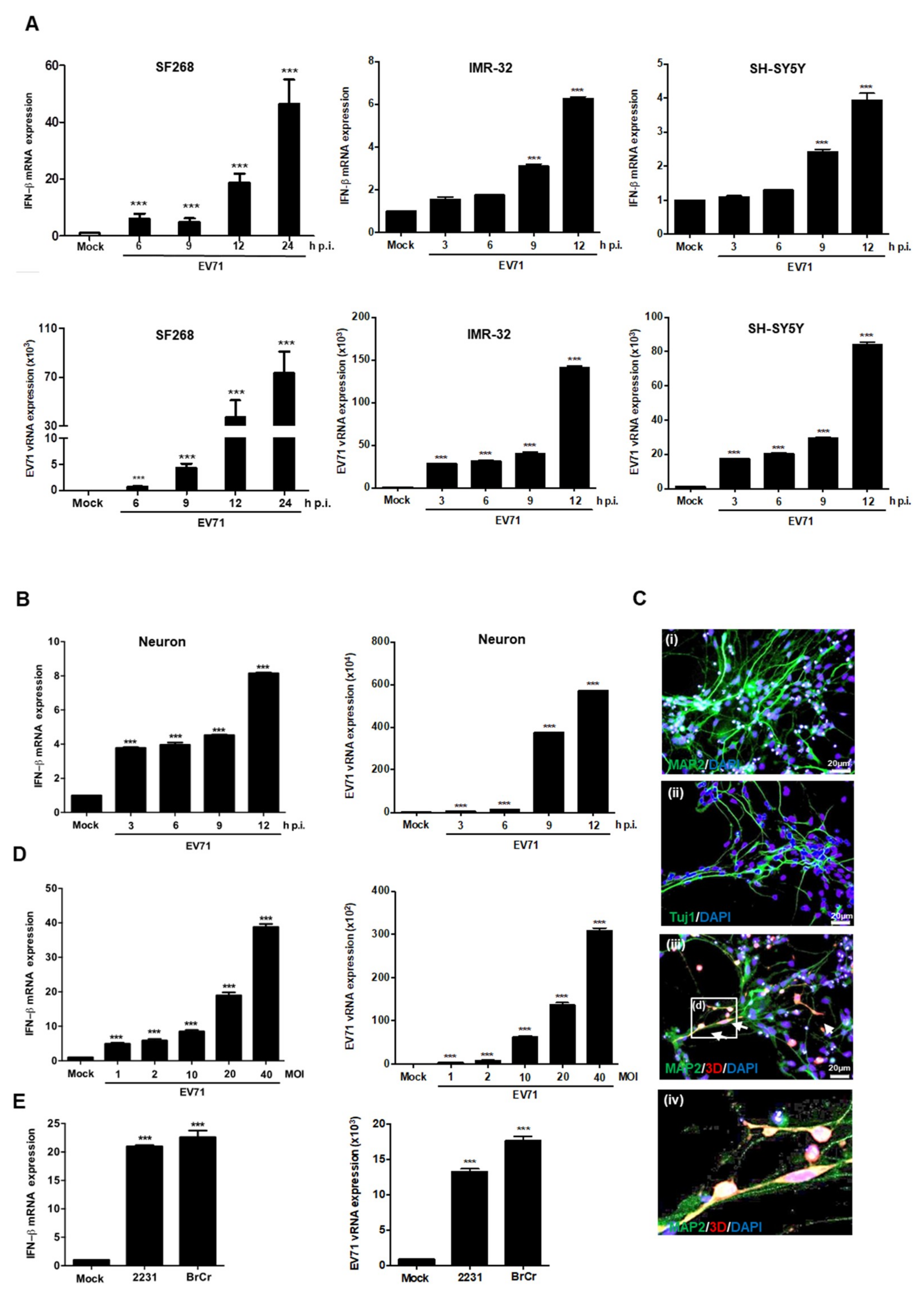
3.1. EV71 Induces IFNβ Expression in Neural Cells
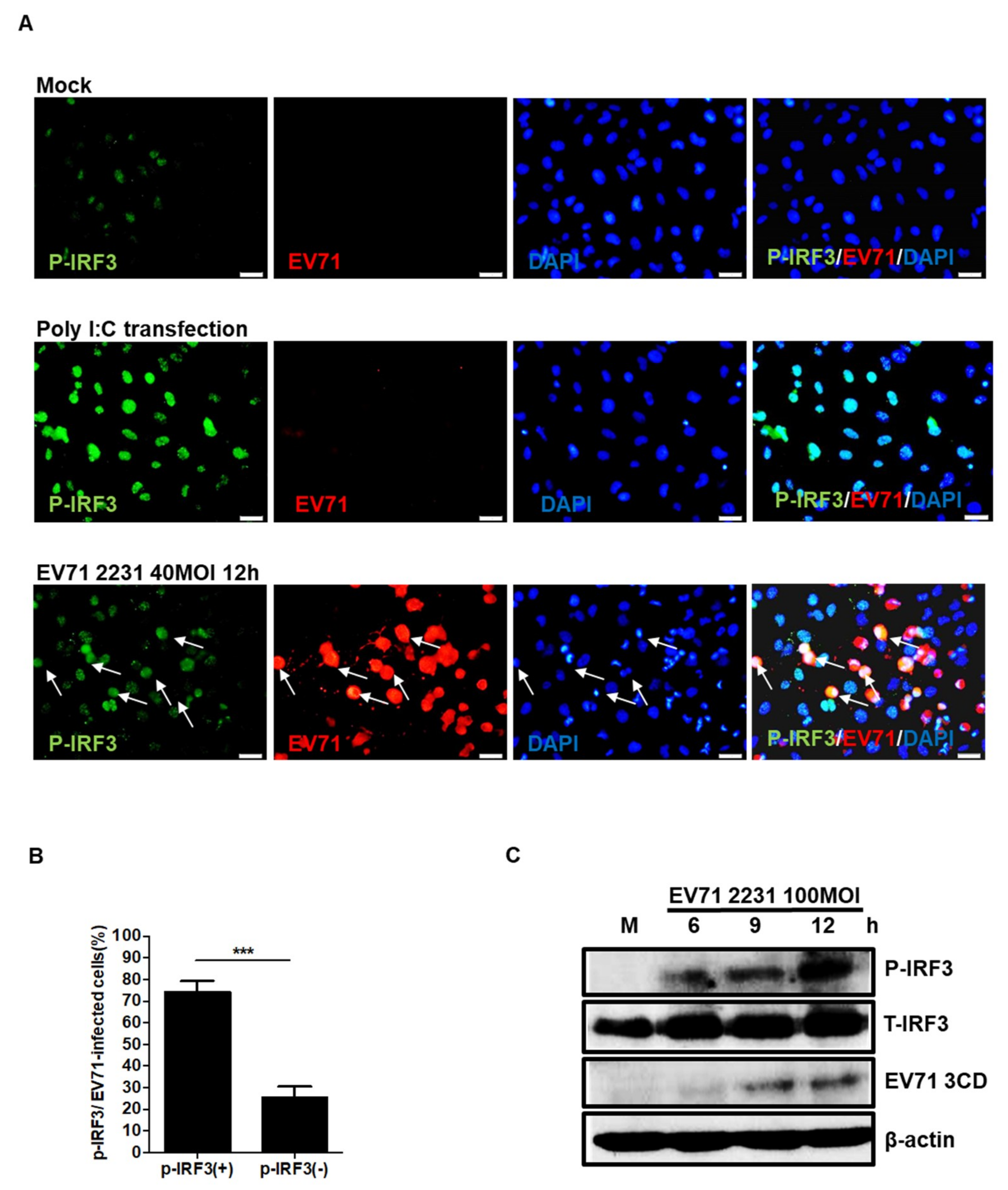
3.2. EV71 Induces IRF3 Phosphorylation in Neuronal SF268 Cells
3.3. EV71 Cleaves TRIF and MAVS in SF268 Cells
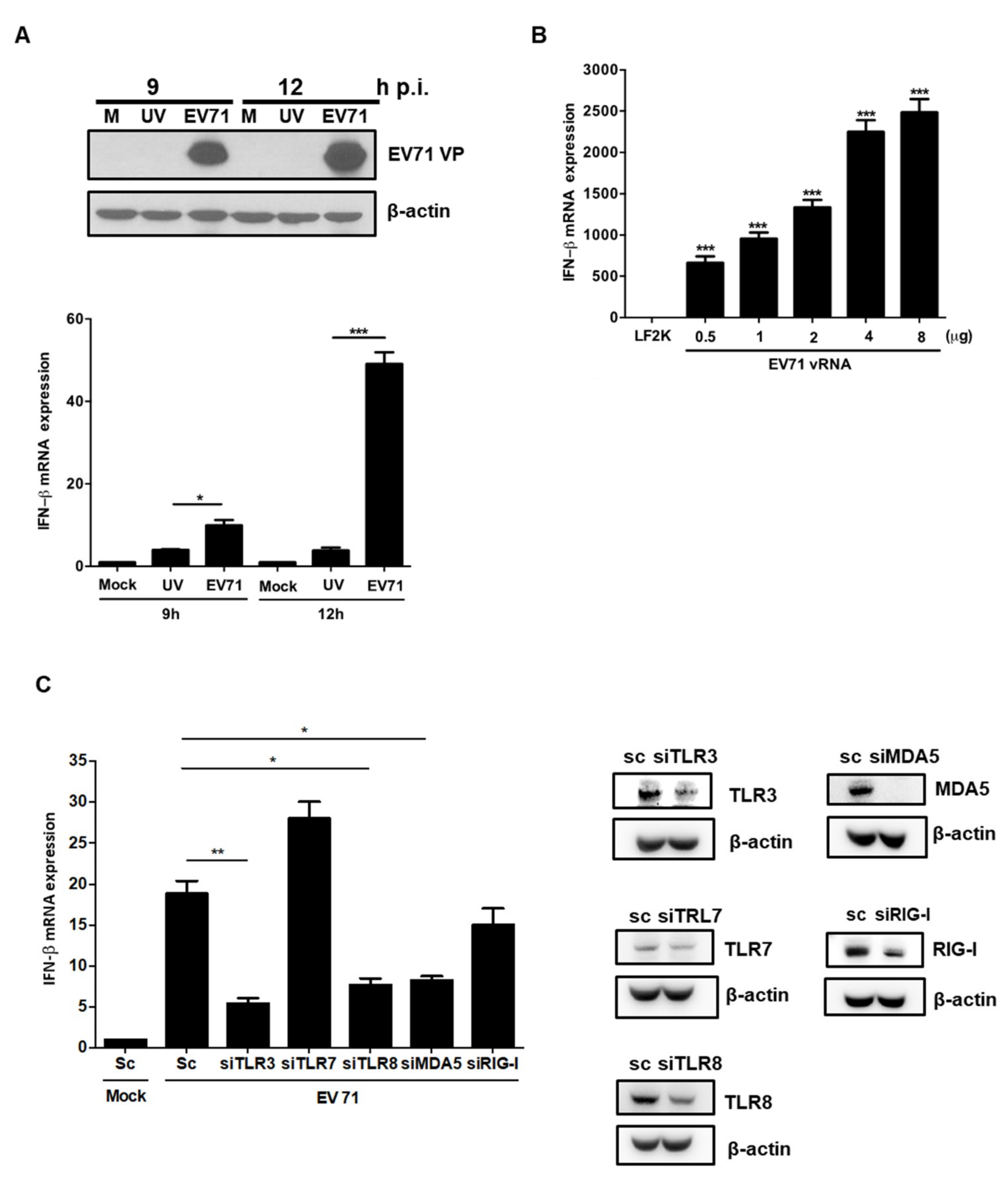
3.4. EV71 RNA Induces IFNβ Expression via TLR3, TLR8, and MDA-5
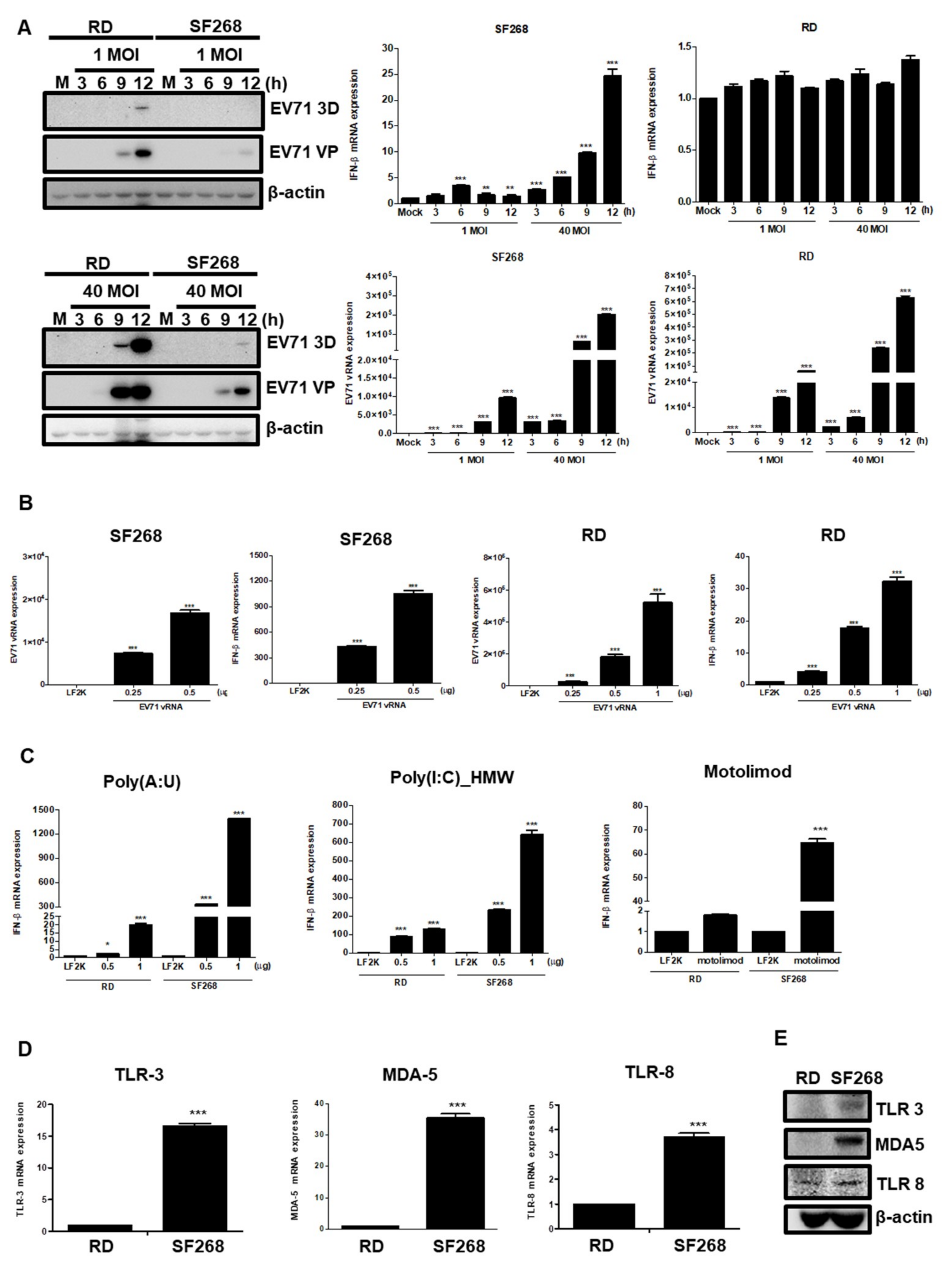
3.5. EV71 vRNA Causes Higher IFNβ Expression in SF268 Cells Than in RD Cells
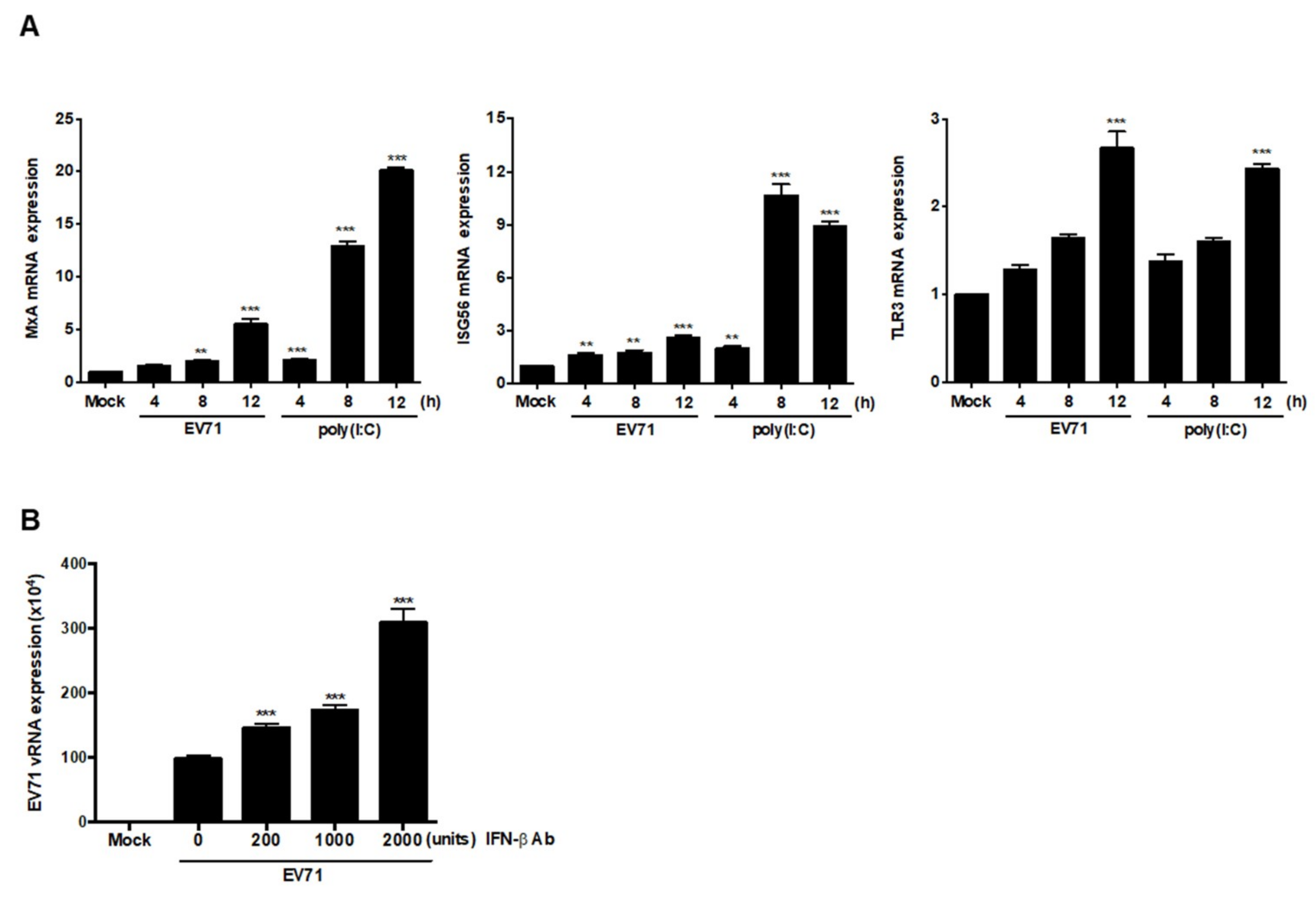
3.6. EV71 Infection Induces Expression of ISGs
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Nagata, N.; Iwasaki, T.; Ami, Y.; Tano, Y.; Harashima, A.; Suzaki, Y.; Sato, Y.; Hasegawa, H.; Sata, T.; Miyamura, T.; et al. Differential localization of neurons susceptible to enterovirus 71 and poliovirus type 1 in the central nervous system of cynomolgus monkeys after intravenous inoculation. J. Gen. Virol. 2004, 85, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Munisamy, B.; Ong, K.C.; Kojima, H.; Noriyo, N.; Chua, K.B.; Ong, B.B.; Nagashima, K. The distribution of inflammation and virus in human enterovirus 71 encephalomyelitis suggests possible viral spread by neural pathways. J. Neuropathol. Exp. Neurol. 2008, 67, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 6, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Müller, M.; Decker, T. The regulation of inflammation by interferons and their STATs. JAK STAT 2013, 2, e23820. [Google Scholar] [CrossRef]
- Koestner, W.; Spanier, J.; Klause, T.; Tegtmeyer, P.K.; Becker, J.; Herder, V.; Borst, K.; Todt, D.; Lienenklaus, S.; Gerhauser, I.; et al. Interferon-beta expression and type I interferon receptor signaling of hepatocytes prevent hepatic necrosis and virus dissemination in Coxsackievirus B3-infected mice. PLoS Pathog. 2018, 14, e1007235. [Google Scholar] [CrossRef]
- Kandolf, R.; Canu, A.; Hofschneider, P.H. Coxsackie B3 virus can replicate in cultured human foetal heart cells and is inhibited by interferon. J. Mol. Cell. Cardiol. 1985, 17, 167–181. [Google Scholar] [CrossRef]
- Rasti, M.; Khanbabaei, H.; Teimoori, A. An update on enterovirus 71 infection and interferon type I response. Rev. Med. Virol. 2019, 29, e2016. [Google Scholar] [CrossRef]
- Lu, J.; Yi, L.; Zhao, J.; Yu, J.; Chen, Y.; Lin, M.C.; Kung, H.F.; He, M.L. Enterovirus 71 Disrupts Interferon Signaling by Reducing the Level of Interferon Receptor 1. J. Virol. 2012, 86, 3767–3776. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Tailor, P.; Tamura, T.; Ozato, K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res. 2006, 16, 134–140. [Google Scholar] [CrossRef]
- Garcı’a-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Watanabe, M.; Goto, H.; Kawaoka, Y. Identification of a Novel Viral Protein Expressed from the PB2 Segment of Influenza A Virus. J. Virol. 2015, 90, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; de Groot, R.J.; van Kuppeveld, F.J.M. Essential Role of Enterovirus 2A Protease in Counteracting Stress Granule Formation and the Induction of Type I Interferon. J. Virol. 2019, 93, e00222–e00319. [Google Scholar] [CrossRef]
- Al-Shujairi, W.H.; Clarke, J.N.; Davies, L.T.; Alsharifi, M.S.M.; Pitson, S.M.; Carr, J.M. Intracranial Injection of Dengue Virus Induces Interferon Stimulated Genes and CD8+ T Cell Infiltration by Sphingosine Kinase 1 Independent Pathways. PLoS ONE 2017, 12, e0169814. [Google Scholar] [CrossRef]
- Rodriguez-Madoz, J.R.; Bernal-Rubio, D.; Kaminski, D.; Boyd, K.; Fernandez-Sesma, A. Dengue Virus Inhibits the Production of Type I Interferon in Primary Human Dendritic Cells. J. Virol. 2010, 84, 4845–4850. [Google Scholar] [CrossRef]
- Mishra, B.B.; Mishra, P.K.; Teale, J.M. Expression and distribution of Toll like receptors in the brain during murine neurocysticercosis. J. Neuroimmunol. 2006, 181, 46–56. [Google Scholar] [CrossRef]
- Delhaye, S.; Paul, S.; Blakqori, G.; Minet, M.; Weber, F.; Staeheli, P.; Michiels, T. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. USA 2006, 103, 7835–7840. [Google Scholar] [CrossRef]
- Préhaud, C.; Mégret, F.; Lafage, M.; Lafon, M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.; Palomo, M.; Alcami, A. Interferon-alpha/beta genes are up-regulated in murine brain astrocytes after infection with Theiler’s murine encephalomyelitis virus. J. Interf. Cytokine Res. 2010, 30, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wang, L.C.; Liu, L.D.; Liao, Y.; Dong, C.H.; Li, Q.H. Biological effects of EV71 infection in neural cells. J. Biophys. Chem. 2010, 2543, 113–118. [Google Scholar] [CrossRef]
- Too, I.H.; Yeo, H.; Sessions, O.M.; Yan, B.; Libau, E.A.; Howe, J.L.; Lim, Z.Q.; Suku-Maran, S.; Ong, W.Y.; Chua, K.B.; et al. Enterovirus 71 infection of motor neuron-like NSC-34 cells undergoes a non-lytic exit pathway. Sci. Rep. 2016, 36983, 1–16. [Google Scholar] [CrossRef]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef]
- Liou, A.T.; Wu, S.Y.; Liao, C.C.; Chang, Y.S.; Chang, C.S.; Shih, C.A. New animal model containing human SCARB2 and lacking stat-1 is highly susceptible to EV71. Sci. Rep. 2016, 31151, 1–10. [Google Scholar] [CrossRef]
- Noah, D.L.; Blum, M.A.; Sherry, B. Interferon regulatory factor 3 is required for viral induction of beta interferon in primary cardiac myocyte cultures. J. Virol. 1999, 73, 10208–10213. [Google Scholar]
- Marsh, B.; Stevens, S.L.; Packard, A.E.; Gopalan, B.; Hunter, B.; Leung, P.Y.; Harrington, C.A.; Stenzel-Poore, M.P. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: A critical role for IRF3. J. Neurosci. 2009, 29, 9839–9849. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef]
- Mohanty, M.C.; Deshpande, J.M. Differential induction of Toll-like receptors & type 1 interferons by Sabin attenuated & wild type 1 polioviruses in human neuronal cells. Indian J. Med. Res. 2013, 138, 209–218. [Google Scholar] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 391, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Hewson, C.A.; Jardine, A.; Edwards, M.R.; Laza-Stanca, V.; Johnston, S.L. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 2005, 79, 12273–12279. [Google Scholar] [CrossRef] [PubMed]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.R.; Yu, C.K.; Kung, S.H.; Chen, S.H.; Chang, C.F.; Ho, T.C.; Lee, Y.P.; Chang, H.C.; Huang, L.Y.; Lo, S.Y.; et al. Toll-Like Receptor 3 Is Involved in Detection of Enterovirus A71 Infection and Targeted by Viral 2A Protease. Viruses 2018, 10, 689. [Google Scholar] [CrossRef]
- Li, B.; Ma, H.M.; Wang, X.X.; Li, Y.Q.; Liu, H.B.; Hong, L.Z.; Li, X.; Zheng, W.H.; Ou, W.L. Expression and significance of toll-like receptors 7 and 8 in brain and lung tissues of death cases caused by EV71 infection. Zhongguo Dang Dai Er Ke Za Zhi 2015, 17, 1051–1055. [Google Scholar]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef]
- Peltier, D.C.; Simms, A.; Farmer, J.R.; Miller, D.J. Human neuronal cells possess functional cytoplasmic and TLR-mediated innate immune pathways influenced by phosphatidylinositol-3 kinase signaling. J. Immunol. 2010, 184, 7010–7021. [Google Scholar] [CrossRef]
- Nazmi, A.; Mukherjee, S.; Kundu, K.; Dutta, K.; Mahadevan, A.; Shankar, S.K.; Basu, A. TLR7 is a key regulator of innate immunity against Japanese encephalitis virus infection. Neurobiol. Dis. 2014, 69, 235–247. [Google Scholar] [CrossRef]
- Co, J.G.; Witwer, K.W.; Gama, L.; Zink, M.C.; Clements, J.E. Induction of Innate Immune Responses by SIV In Vivo and In Vitro: Differential Expression and Function of RIG-I and MDA5. J. Infect. Dis. 2011, 204, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.L.; Kao, L.T.; Lin, S.J.; Wang, R.Y.; Shih, S.R. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS ONE 2013, 8, e63431. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.G.; Knowell, A.E.; Hunt, A.; Patel, D.; Bhosle, S.; Chaudhary, J. Interferon inducible antiviral MxA is inversely associated with prostate cancer and regulates cell cycle, invasion and Docetaxel induced apoptosis. Prostate 2015, 75, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Zhang, L.Y.; Wang, Z.Y.; Wang, Y.; Song, F.M.; Zhang, Y.H.; Yu, J.H. Rheum emodin inhibits enterovirus 71 viral replication and affects the host cell cycle environment. Acta Pharmacol. Sin. 2017, 38, 392–401. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Sequence (5′–3′) | |
|---|---|---|
| EV71 5′UTR | Forward | CCC TGA ATG CGG CTA ATC C |
| Reverse | ATT GTC ACC ATA AGC AGC CA | |
| Human IFN | Forward | AGA AGG AGG ACG CCG CAT TG |
| Reverse | TCA GTT TCG GAG GTA ACC TG | |
| Human TLR3 | Forward | AAG GGT GGC CCT TAA AAA TG |
| Reverse | GTT TCC AGA GCC GTG CTA AG | |
| Human TLR8 | Forward | CAG AGC ATC AAC CAA AGC AA |
| Reverse | GCT GCC GTA GCC TCA AAT AC | |
| Human MDA5 | Forward | AGG AGT CAA AGC CCA CCA TCT G |
| Reverse | ATT GGT GAC GAG ACC ATA ACG GAT A | |
| Human ISG56 | Forward | TCT CAG AGG AGC CTG GCT AAG |
| Reverse | CCA CAC TGT ATT TGG TGT CTA GG | |
| Human MxA | Forward | TTC AGC ACC TGA TGG CCT ATC |
| Reverse | TGG ATG ATC AAA GGG ATG TGG | |
| Human 18S rRNA | Forward | GTA ACC CGT TGA ACC CCA TT |
| Reverse | CCA TCC AAT CGG TAG TAG CG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.-I.; Lin, J.-Y.; Chen, S.-H. EV71 Infection Induces IFNβ Expression in Neural Cells. Viruses 2019, 11, 1121. https://doi.org/10.3390/v11121121
Huang H-I, Lin J-Y, Chen S-H. EV71 Infection Induces IFNβ Expression in Neural Cells. Viruses. 2019; 11(12):1121. https://doi.org/10.3390/v11121121
Chicago/Turabian StyleHuang, Hsing-I, Jhao-Yin Lin, and Sheng-Hung Chen. 2019. "EV71 Infection Induces IFNβ Expression in Neural Cells" Viruses 11, no. 12: 1121. https://doi.org/10.3390/v11121121
APA StyleHuang, H.-I., Lin, J.-Y., & Chen, S.-H. (2019). EV71 Infection Induces IFNβ Expression in Neural Cells. Viruses, 11(12), 1121. https://doi.org/10.3390/v11121121