ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage

 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals
2.3. Isolation of Nucleic Acids
2.4. PCR Analysis
2.5. Knockdown of DDR Factors
2.6. ATM and ATR Overexpression
2.7. Immunocytochemistry
2.8. Quantification of HBsAg
2.9. Statistical Analysis
3. Results
3.1. DNA Damage Promotes HBV Replication
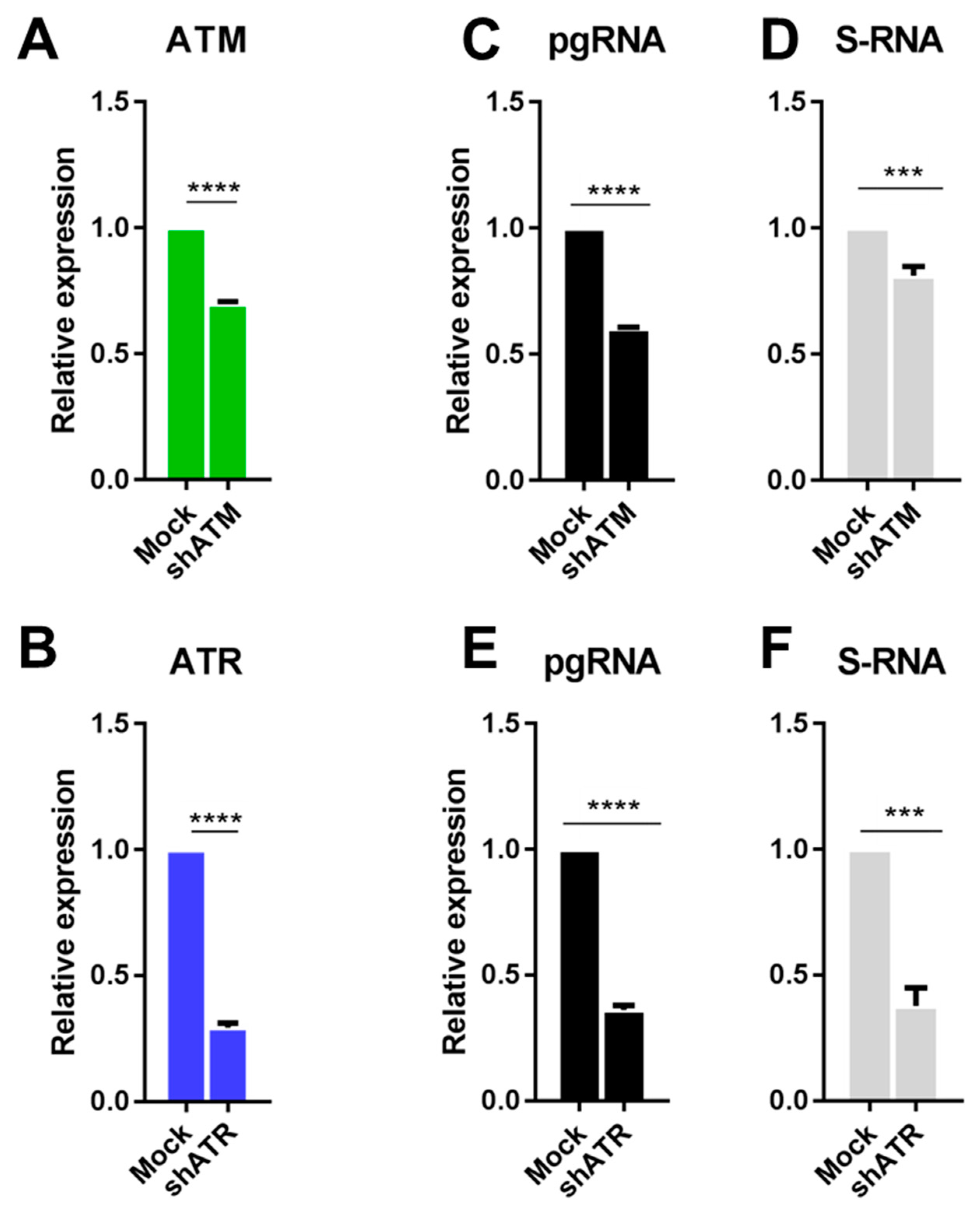
3.2. The Role of ATM and ATR in HBV Replication
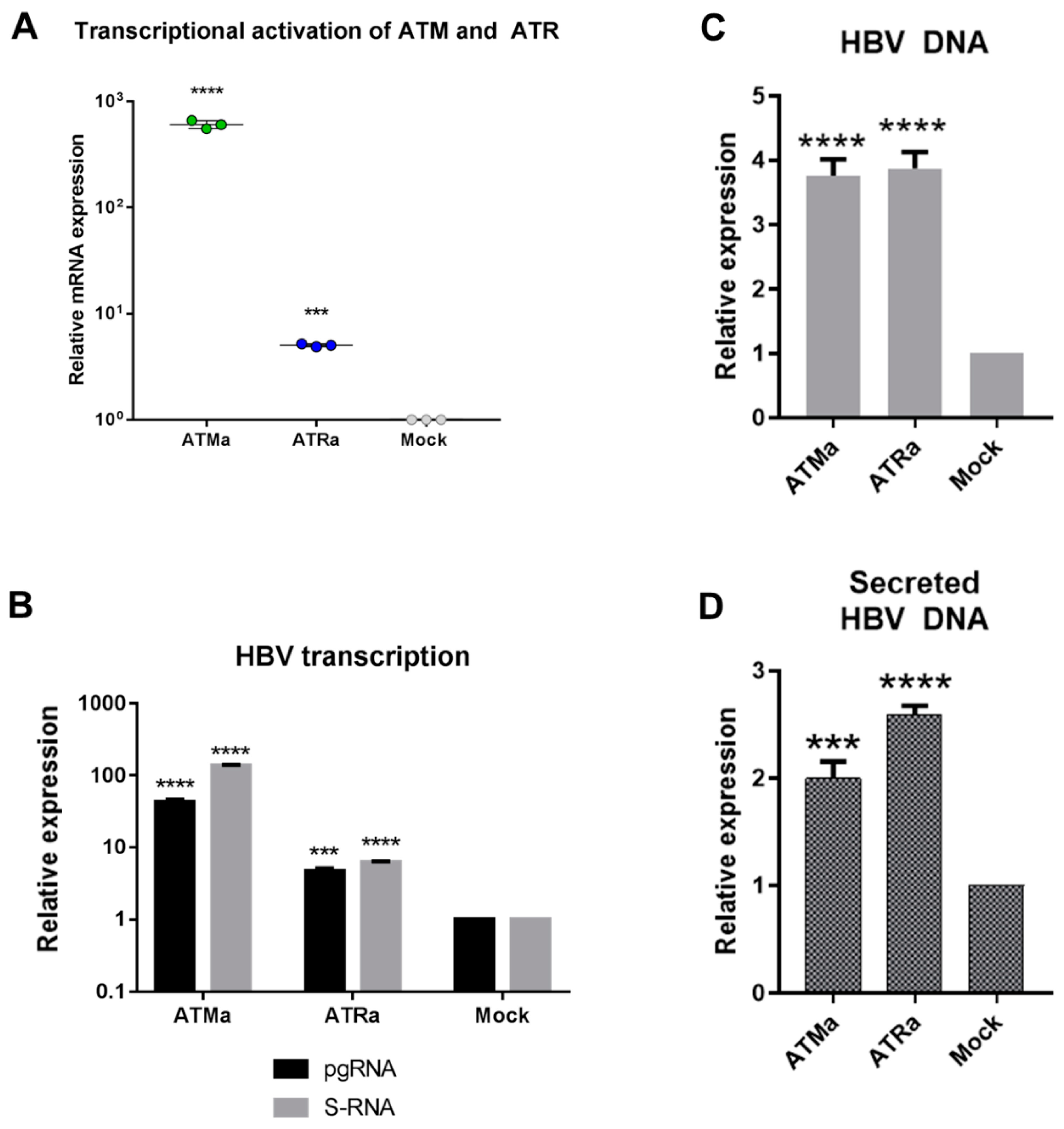
3.3. Transcriptional Activation of ATM and ATR Promotes HBV Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seto, W.-K.; Lo, Y.-R.; Pawlotsky, J.-M.; Yuen, M.-F. Chronic Hepatitis B Virus Infection. Lancet 2018, 392, 2313–2324. [Google Scholar] [CrossRef]
- World Health Organization. Global Hepatitis Report, 2017; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-156545-5.
- Lampertico, P.; Invernizzi, F.; Vigano, M.; Loglio, A.; Mangia, G.; Facchetti, F.; Primignani, M.; Jovani, M.; Iavarone, M.; Fraquelli, M.; et al. The Long-Term Benefits of Nucleos(t)Ide Analogs in Compensated HBV Cirrhotic Patients with No or Small Esophageal Varices: A 12-Year Prospective Cohort Study. J. Hepatol. 2015, 63, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Liang, R.H.; Chiu, E.K.; Wong, K.L.; Chan, T.K.; Todd, D. Reactivation of Hepatitis B Virus Replication in Patients Receiving Cytotoxic Therapy. Report of a Prospective Study. Gastroenterology 1991, 100, 182–188. [Google Scholar] [CrossRef]
- Loomba, R.; Liang, T.J. Hepatitis B Reactivation Associated With Immune Suppressive and Biological Modifier Therapies: Current Concepts, Management Strategies, and Future Directions. Gastroenterology 2017, 152, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.A.; Perrillo, R.P. Hepatitis B Virus Reactivation in the Setting of Cancer Chemotherapy and Other Immunosuppressive Drug Therapy. Clin. Infect. Dis. 2016, 62, S306–S313. [Google Scholar] [CrossRef] [PubMed]
- Perceau, G.; Diris, N.; Estines, O.; Derancourt, C.; Levy, S.; Bernard, P. Late Lethal Hepatitis B Virus Reactivation after Rituximab Treatment of Low-Grade Cutaneous B-Cell Lymphoma. Br. J. Dermatol. 2006, 155, 1053–1056. [Google Scholar] [CrossRef]
- Matsumoto, T.; Marusawa, H.; Dogaki, M.; Suginoshita, Y.; Inokuma, T. Adalimumab-Induced Lethal Hepatitis B Virus Reactivation in an HBsAg-Negative Patient with Clinically Resolved Hepatitis B Virus Infection. Liver Int. 2010, 30, 1241–1242. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Hsu, H.-C.; Chen, H.-L.; Gao, M.; Yeh, P.-Y.; Chen, P.-J.; Cheng, A.-L. Doxorubicin Activates Hepatitis B Virus (HBV) Replication in HBV-Harboring Hepatoblastoma Cells. A Possible Novel Mechanism of HBV Reactivation in HBV Carriers Receiving Systemic Chemotherapy. Anticancer Res. 2004, 24, 3035–3040. [Google Scholar]
- Wang, J.; Jia, J.; Chen, R.; Ding, S.; Xu, Q.; Zhang, T.; Chen, X.; Liu, S.; Lu, F. RFX1 Participates in Doxorubicin-Induced Hepatitis B Virus Reactivation. Cancer Med. 2018, 7, 2021–2033. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Chong, C.-L.; Wu, Y.-C.; Wang, Y.-L.; Tsai, K.-N.; Kuo, T.-M.; Hong, M.-H.; Hu, C.-P.; Chen, M.-L.; Chou, Y.-C.; et al. Doxorubicin Activates Hepatitis B Virus Replication by Elevation of P21 (Waf1/Cip1) and C/EBPα Expression. PLoS ONE 2015, 10, e0131743. [Google Scholar] [CrossRef]
- Xu, L.; Tu, Z.; Xu, G.; Hu, J.-L.; Cai, X.-F.; Zhan, X.-X.; Wang, Y.-W.; Huang, Y.; Chen, J.; Huang, A.-L. S-Phase Arrest after Vincristine Treatment May Promote Hepatitis B Virus Replication. World J. Gastroenterol. 2015, 21, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, E.; Zhu, J.; Xu, L.; Chen, X.; Li, J.; Liang, L.; Hu, Y.; Xia, J.; Chen, J.; et al. Cisplatin Enhances Hepatitis B Virus Replication and PGC-1α Expression through Endoplasmic Reticulum Stress. Sci. Rep. 2018, 8, 3496. [Google Scholar] [CrossRef]
- Gómez-Moreno, A.; Garaigorta, U. Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection. Viruses 2017, 9, 304. [Google Scholar] [CrossRef]
- Chung, Y.-L.; Tsai, T.-Y. Promyelocytic Leukemia Nuclear Bodies Link the DNA Damage Repair Pathway with Hepatitis B Virus Replication: Implications for Hepatitis B Virus Exacerbation during Chemotherapy and Radiotherapy. Mol. Cancer Res. 2009, 7, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR Signaling at a Glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef]
- Panda, S.; Isbatan, A.; Adami, G.R. Modification of the ATM/ATR Directed DNA Damage Response State with Aging and Long after Hepatocyte Senescence Induction in Vivo. Mech. Ageing Dev. 2008, 129, 332–340. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Turnell, A.S.; Grand, R.J. DNA Viruses and the Cellular DNA-Damage Response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Kostyushev, D.S.; Brezgin, S.A.; Kostyusheva, A.P.; Lipatnikov, A.D.; Simirskii, V.N.; Mamonova, N.A.; Volchkova, E.V.; Maleyev, V.V.; Chulanov, V. Increased Formation of Phosphorylated H2AX Foci in Nuclei of Cells Infected by Hepatitis B AND B+D Viruses. Vopr. Virusol. 2018, 63, 165–170. [Google Scholar]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Osawa, M.; Takamura, M.; Yamagiwa, S.; Aoyagi, Y.; Sanpei, A.; Fujimaki, S. DNA Damage Sensor γ-H2AX Is Increased in Preneoplastic Lesions of Hepatocellular Carcinoma. Sci. World J. 2013, 2013, 597095. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Giachino, C. Multiple Facets of Histone Variant H2AX: A DNA Double-Strand-Break Marker with Several Biological Functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Hou, N.-B.; Song, T.; He, X.; Zheng, Z.-R.; Ma, Q.-J.; Li, L.; Zhang, Y.-H.; Zhong, H. Cellular DNA Repair Cofactors Affecting Hepatitis B Virus Infection and Replication. World J. Gastroenterol. 2008, 14, 5059–5065. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.-S.; Ji, J.-H.; Cho, M.-Y.; Yoo, Y.-S.; Park, Y.-Y.; Cha, H.-J.; Lee, Y.; Kim, Y.; Cho, H. Hepatitis B Virus X Protein Activates the ATM-Chk2 Pathway and Delays Cell Cycle Progression. J. Gen. Virol. 2015, 96, 2242–2251. [Google Scholar] [CrossRef]
- Woods, D.; Turchi, J.J. Chemotherapy Induced DNA Damage Response: Convergence of Drugs and Pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef]
- Driessens, N.; Versteyhe, S.; Ghaddhab, C.; Burniat, A.; De Deken, X.; Van Sande, J.; Dumont, J.-E.; Miot, F.; Corvilain, B. Hydrogen Peroxide Induces DNA Single- and Double-Strand Breaks in Thyroid Cells and Is Therefore a Potential Mutagen for This Organ. Endocr. Relat. Cancer 2009, 16, 845–856. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Kostyusheva, A.P.; Kostyushev, D.S.; Brezgin, S.A.; Zarifyan, D.N.; Volchkova, E.V.; Chulanov, V.P. Small Molecular Inhibitors of DNA Double Strand Break Repair Pathways Increase the ANTI-HBV Activity of CRISPR/Cas9. Mol. Biol. 2019, 53, 274–285. [Google Scholar] [CrossRef]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Zarifyan, D.; Utkina, A.; Goptar, I.; Chulanov, V. Suppressing the NHEJ Pathway by DNA-PKcs Inhibitor NU7026 Prevents Degradation of HBV CccDNA Cleaved by CRISPR/Cas9. Sci. Rep. 2019, 9, 1847. [Google Scholar] [CrossRef]
- Nassal, M. The Arginine-Rich Domain of the Hepatitis B Virus Core Protein Is Required for Pregenome Encapsidation and Productive Viral Positive-Strand DNA Synthesis but Not for Virus Assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [PubMed]
- Li, F.; Cheng, L.; Murphy, C.M.; Reszka-Blanco, N.J.; Wu, Y.; Chi, L.; Hu, J.; Su, L. Minicircle HBV CccDNA with a Gaussia Luciferase Reporter for Investigating HBV CccDNA Biology and Developing CccDNA-Targeting Drugs. Sci. Rep. 2016, 6, 36483. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Nie, H.; Yan, R.; Guo, J.-T.; Block, T.M.; Guo, H. A Southern Blot Assay for Detection of Hepatitis B Virus Covalently Closed Circular DNA from Cell Cultures. In Antiviral Methods and Protocols. Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2013; Volume 1030, pp. 151–161. [Google Scholar]
- Larsson, S.B.; Malmström, S.; Hannoun, C.; Norkrans, G.; Lindh, M. Mechanisms Downstream of Reverse Transcription Reduce Serum Levels of HBV DNA but Not of HBsAg in Chronic Hepatitis B Virus Infection. Virol. J. 2015, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M.; Davies, A.A.; West, S.C. RAD51 Localization and Activation Following DNA Damage. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H.J. The MRE11 Complex: Starting from the Ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ichida, T. Impact of Hepatitis B Virus X Protein on the DNA Damage Response during Hepatocarcinogenesis. Med. Mol. Morphol. 2009, 42, 138–142. [Google Scholar] [CrossRef]
- Kew, M.C. Hepatitis B Virus x Protein in the Pathogenesis of Hepatitis B Virus-Induced Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 144–152. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress, a Trigger of Hepatitis C and B Virus-Induced Liver Carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef]
- Capovilla, A.; Carmona, S.; Arbuthnot, P. Hepatitis B Virus X-Protein Binds Damaged DNA and Sensitizes Liver Cells to Ultraviolet Irradiation. Biochem. Biophys. Res. Commun. 1997, 232, 255–260. [Google Scholar] [CrossRef]
- Jia, L.; Wang, X.W.; Harris, C.C. Hepatitis B Virus X Protein Inhibits Nucleotide Excision Repair. Int. J. Cancer 1999, 80, 875–879. [Google Scholar] [CrossRef]
- Becker, S.A.; Lee, T.-H.; Butel, J.S.; Slagle, B.L. Hepatitis B Virus X Protein Interferes with Cellular DNA Repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [PubMed]
- Schreiner, S.; Nassal, M. A Role for the Host DNA Damage Response in Hepatitis B Virus CccDNA Formation—and Beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef]
- Narayanan, I.V.; Paulsen, M.T.; Bedi, K.; Berg, N.; Ljungman, E.A.; Francia, S.; Veloso, A.; Magnuson, B.; di Fagagna, F.D.A.; Wilson, T.E.; et al. Transcriptional and Post-Transcriptional Regulation of the Ionizing Radiation Response by ATM and P53. Sci. Rep. 2017, 7, 43598. [Google Scholar] [CrossRef]
- Rashi-Elkeles, S.; Warnatz, H.-J.; Elkon, R.; Kupershtein, A.; Chobod, Y.; Paz, A.; Amstislavskiy, V.; Sultan, M.; Safer, H.; Nietfeld, W.; et al. Parallel Profiling of the Transcriptome, Cistrome, and Epigenome in the Cellular Response to Ionizing Radiation. Sci. Signal. 2014, 7, rs3. [Google Scholar] [CrossRef]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone Deacetylase Regulation of ATM-Mediated DNA Damage Signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef]
- Wickramasinghe, V.O.; Venkitaraman, A.R. RNA Processing and Genome Stability: Cause and Consequence. Mol. Cell 2016, 61, 496–505. [Google Scholar] [CrossRef]
- Burger, K.; Ketley, R.F.; Gullerova, M. Beyond the Trinity of ATM, ATR, and DNA-PK: Multiple Kinases Shape the DNA Damage Response in Concert With RNA Metabolism. Front. Mol. Biosci. 2019, 6, 61. [Google Scholar] [CrossRef]
- Li, J.; Hart, R.P.; Mallimo, E.M.; Swerdel, M.R.; Kusnecov, A.W.; Herrup, K. EZH2-Mediated H3K27 Trimethylation Mediates Neurodegeneration in Ataxia-Telangiectasia. Nat. Neurosci. 2013, 16, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA Repair Proteins Affect the Lifecycle of Herpes Simplex Virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, O.; Donovan, K.; Azizkhan-Clifford, J. Inhibition of Ataxia Telangiectasia Mutated (ATM) Kinase Suppresses Herpes Simplex Virus Type 1 (HSV-1) Keratitis. Invest. Ophthalmol. Vis. Sci. 2014, 55, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. MBio 2018, 9, e00064-18. [Google Scholar] [CrossRef]
- Jiang, M.; Zhao, L.; Gamez, M.; Imperiale, M.J. Roles of ATM and ATR-Mediated DNA Damage Responses during Lytic BK Polyomavirus Infection. PLoS Pathog. 2012, 8, e1002898. [Google Scholar] [CrossRef]
- Dahl, J.; You, J.; Benjamin, T.L. Induction and Utilization of an ATM Signaling Pathway by Polyomavirus. J. Virol. 2005, 79, 13007–13017. [Google Scholar] [CrossRef]
- Anastasiou, O.E.; Theissen, M.; Verheyen, J.; Bleekmann, B.; Wedemeyer, H.; Widera, M.; Ciesek, S. Clinical and Virological Aspects of HBV Reactivation: A Focus on Acute Liver Failure. Viruses 2019, 11, 863. [Google Scholar] [CrossRef]
- Pisaturo, M.; Macera, M.; Alessio, L.; Calo, F.; Coppola, N. Hepatitis B Virus (HBV) Reactivation Following Pharmacological Eradication of Hepatitis C Virus (HCV). Viruses 2019, 11, 850. [Google Scholar] [CrossRef]
- Inoue, J.; Nakamura, T.; Masamune, A. Roles of Hepatitis B Virus Mutations in the Viral Reactivation after Immunosuppression Therapies. Viruses 2019, 11, 457. [Google Scholar] [CrossRef]
- Bersoff-Matcha, S.J.; Cao, K.; Jason, M.; Ajao, A.; Jones, S.C.; Meyer, T.; Brinker, A. Hepatitis B Virus Reactivation Associated With Direct-Acting Antiviral Therapy for Chronic Hepatitis C Virus: A Review of Cases Reported to the U.S. Food and Drug Administration Adverse Event Reporting System. Ann. Intern. Med. 2017, 166, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Bozza, C.; Cinausero, M.; Iacono, D.; Puglisi, F. Hepatitis B and Cancer: A Practical Guide for the Oncologist. Crit. Rev. Oncol. Hematol. 2016, 98, 137–146. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Number | Concentration (mM) |
| H2O2 | 1 | 0.01 |
| 2 | 0.4 | |
| 3 | 2 | |
| 4 | 3 | |
| 5 | 4 | |
| 6 | 7 | |
| 7 | 8 | |
| 8 | 9 | |
| Agent | Number | Concentration (μM) |
| Doxorubicin | 1 | 0.1 |
| 2 | 0.2 | |
| 3 | 1.5 |
| Target | Sequence | |
|---|---|---|
| GAPDH mRNA | fw | 5′-CCAGGTGGTCTCCTCTGACTT-3′ |
| rev | 5′-GTTGCTGTAGCCAAATTCGTTGT-3′ | |
| probe | FAM-AACAGCGACACCCACTCCTCCACC-BHQ1 | |
| cссDNA | fw | 5′-CCGTGTGCACTTCGCTTCA-3′ |
| rev | 5′-GCACAGCTTGGAGGCTTGA-3′ | |
| probe | FAM-CATGGAGACCACCGTGAACGCCC-BHQ1 | |
| pgRNA | fw | 5′-GGTCCCCTAGAAGAAGAACTCCCT-3′ [35] |
| rev | 5′-CATTGAGATTCCCGAGATTGAGAT-3′ [35] | |
| probe | FAM-TCTCAATCGCCGCGTCGCAGA-BHQ1 [35] | |
| S-RNA | fw | 5′-TCCTCCAAСTTGTCCTGGTTATC-3′ [35] |
| rev | 5′-AGATGAGGCATAGCAGCAGGAT-3′ [35] | |
| probe | FAM-ATGATAAAACGCCGCAGACACATCCAGC-BHQ1 [35] | |
| HBV DNA and β-globin | АmpliSens® HBV monitor (CRIE) | |
| ATM mRNA | fw | 5′-AGTTTCATCTTCCGGCCTCT-3′ |
| rev | 5′-GCTGTGAGAAAACCATGGAAG-3′ | |
| ATR mRNA | fw | 5′-AACATTCGTGGCATTGACTG-3′ |
| rev | 5′-AAGCAAGGTGATCTCATCCG-3′ | |
| PRKDC mRNA | fw | 5′-CTTACATGCTAATGTATAAGGGCG-3′ |
| rev | 5′-CAGCAGGCACTTTACTTTCTC-3′ | |
| MRE11A mRNA | fw | 5′-TCAGTTAGGTGGGTCTGGGT-3′ |
| rev | 5′-AGCGGTGAACTGAATCGCAT-3′ | |
| RAD51 mRNA | fw | 5′-TCACGGTTAGAGCAGTGTG-3′ |
| rev | 5′-AACAGCCTCCACAGTATGG-3′ | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostyusheva, A.; Brezgin, S.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Urusov, F.; Nikiforova, A.; Volchkova, E.; Kostyushev, D.; et al. ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage. Viruses 2019, 11, 997. https://doi.org/10.3390/v11110997
Kostyusheva A, Brezgin S, Bayurova E, Gordeychuk I, Isaguliants M, Goptar I, Urusov F, Nikiforova A, Volchkova E, Kostyushev D, et al. ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage. Viruses. 2019; 11(11):997. https://doi.org/10.3390/v11110997
Chicago/Turabian StyleKostyusheva, Anastasiya, Sergey Brezgin, Ekaterina Bayurova, Ilya Gordeychuk, Maria Isaguliants, Irina Goptar, Felix Urusov, Anastasiya Nikiforova, Elena Volchkova, Dmitry Kostyushev, and et al. 2019. "ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage" Viruses 11, no. 11: 997. https://doi.org/10.3390/v11110997
APA StyleKostyusheva, A., Brezgin, S., Bayurova, E., Gordeychuk, I., Isaguliants, M., Goptar, I., Urusov, F., Nikiforova, A., Volchkova, E., Kostyushev, D., & Chulanov, V. (2019). ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage. Viruses, 11(11), 997. https://doi.org/10.3390/v11110997