RETRACTED: Contribution of Host Immune Responses against Influenza D Virus Infection toward Secondary Bacterial Infection in a Mouse Model

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Virus Preparation
2.3. Mice
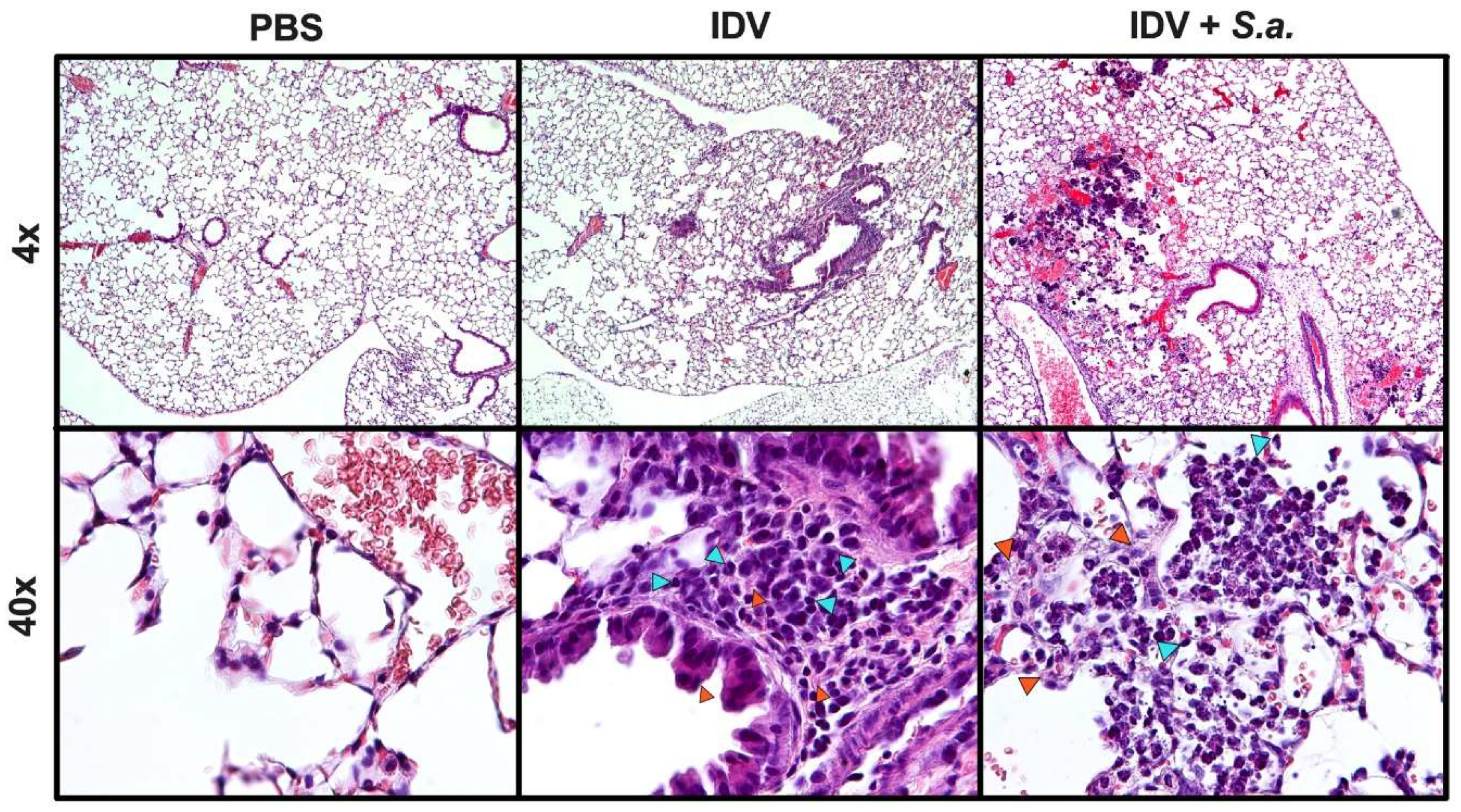
2.4. Mouse Inoculations, Challenge, Burden, Morbidity, Histology, and Survival
2.5. Preparation of BALF Samples and Cell Population Analysis
2.6. A549 Cell Infections
2.7. RNA Isolation
2.8. One-Step RT-qPCR
2.9. ELISA
2.10. Statistical Analysis
3. Results
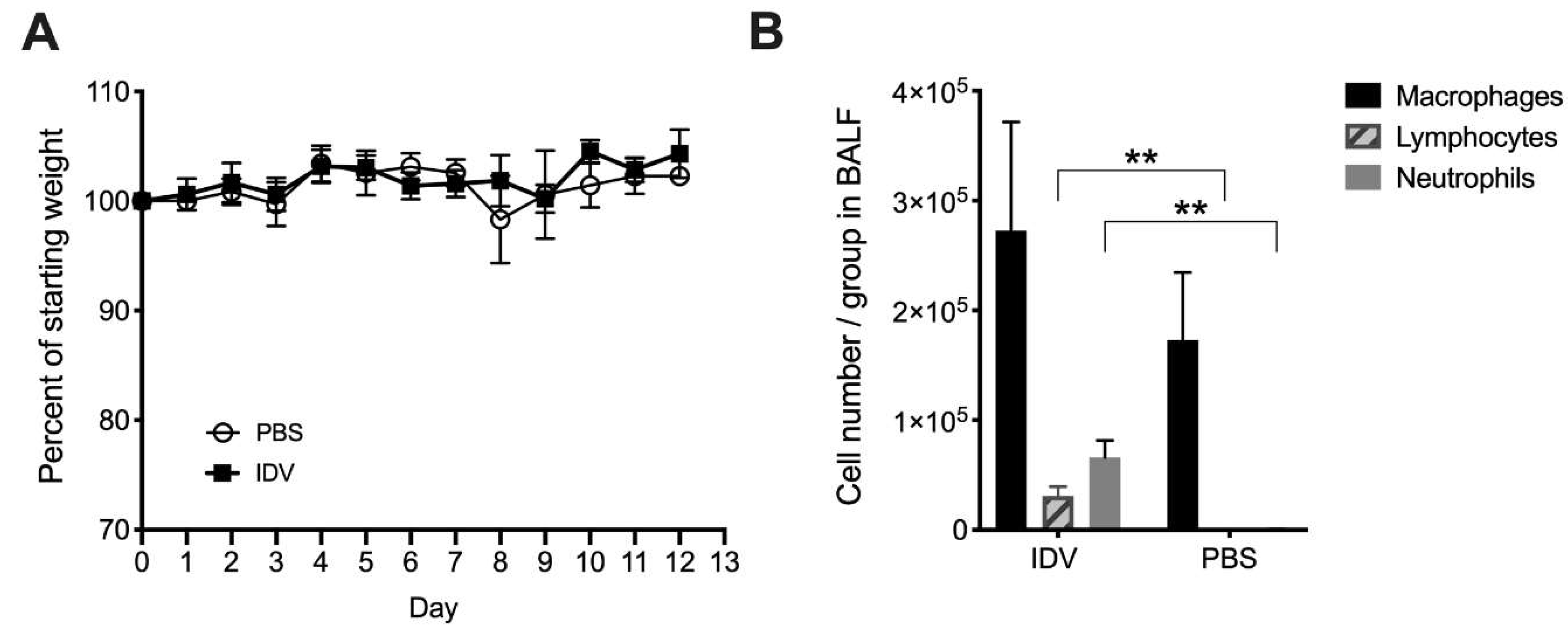
3.1. IDV Infection in C57BL/6 Mice is Asymptomatic
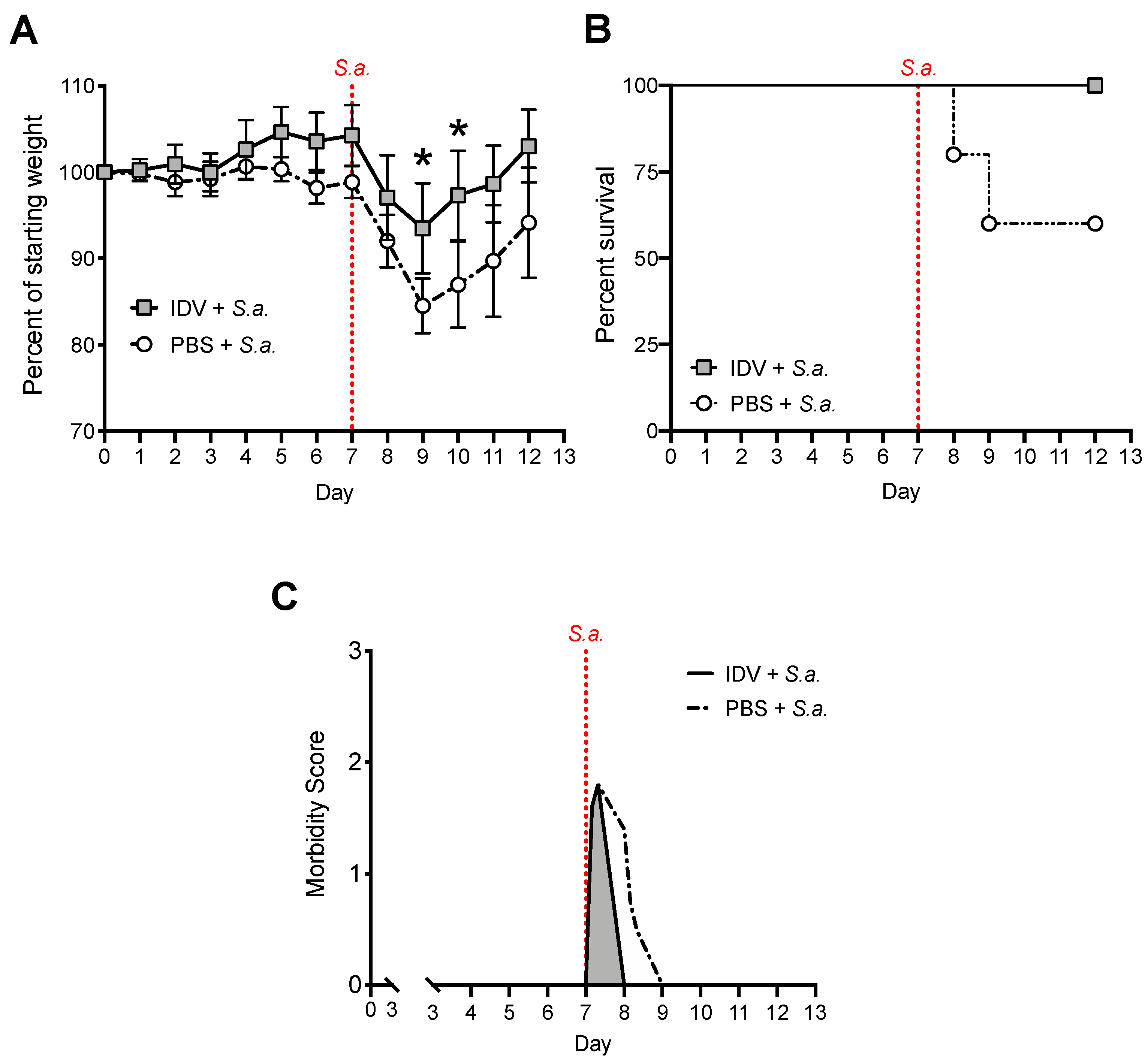
3.2. IDV Infection Improves Survival after S. aureus Secondary Infection but Does Not Affect Bacterial Clearance
3.3. IDV Enhances IFN-β Expression by Lung Epithelial Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Asha, K.; Kumar, B. Emerging influenza d virus threat: What we know so far! J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and swine: Proposal for a new genus in the orthomyxoviridae family. mBio 2014, 5, e00031-14. [Google Scholar] [CrossRef]
- Ferguson, L.; Olivier, A.K.; Genova, S.; Epperson, W.B.; Smith, D.R.; Schneider, L.; Barton, K.; McCuan, K.; Webby, R.J.; Wan, X.F. Pathogenesis of influenza d virus in cattle. J. Virol. 2016, 90, 5636–5642. [Google Scholar] [CrossRef]
- Luo, J.; Ferguson, L.; Smith, D.R.; Woolums, A.R.; Epperson, W.B.; Wan, X.F. Serological evidence for high prevalence of influenza d viruses in cattle, nebraska, united states, 2003-2004. Virology 2017, 501, 88–91. [Google Scholar] [CrossRef]
- Gatherer, D. Tempo and mode in the molecular evolution of influenza c. PLoS Curr 2010, 2, RRN1199. [Google Scholar] [CrossRef]
- Su, S.; Fu, X.; Li, G.; Kerlin, F.; Veit, M. Novel influenza d virus: Epidemiology, pathology, evolution and biological characteristics. Virulence 2017, 8, 1580–1591. [Google Scholar] [CrossRef]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a novel swine influenza virus from oklahoma in 2011 which is distantly related to human influenza c viruses. PLoS Pathog 2013, 9, e1003176. [Google Scholar] [CrossRef]
- Mitra, N.; Cernicchiaro, N.; Torres, S.; Li, F.; Hause, B.M. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza d virus. J. Gen. Virol 2016, 97, 1771–1784. [Google Scholar] [CrossRef]
- Chiapponi, C.; Faccini, S.; De Mattia, A.; Baioni, L.; Barbieri, I.; Rosignoli, C.; Nigrelli, A.; Foni, E. Detection of influenza d virus among swine and cattle, italy. Emerg Infect. Dis 2016, 22, 352–354. [Google Scholar] [CrossRef]
- Ducatez, M.F.; Pelletier, C.; Meyer, G. Influenza d virus in cattle, france, 2011-2014. Emerg Infect. Dis 2015, 21, 368–371. [Google Scholar] [CrossRef]
- Flynn, O.; Gallagher, C.; Mooney, J.; Irvine, C.; Ducatez, M.; Hause, B.; McGrath, G.; Ryan, E. Influenza d virus in cattle, ireland. Emerg Infect. Dis 2018, 24, 389–391. [Google Scholar] [CrossRef]
- Murakami, S.; Endoh, M.; Kobayashi, T.; Takenaka-Uema, A.; Chambers, J.K.; Uchida, K.; Nishihara, M.; Hause, B.; Horimoto, T. Influenza d virus infection in herd of cattle, japan. Emerg Infect. Dis 2016, 22, 1517–1519. [Google Scholar] [CrossRef]
- Quast, M.; Sreenivasan, C.; Sexton, G.; Nedland, H.; Singrey, A.; Fawcett, L.; Miller, G.; Lauer, D.; Voss, S.; Pollock, S.; et al. Serological evidence for the presence of influenza d virus in small ruminants. Vet. Microbiol 2015, 180, 281–285. [Google Scholar] [CrossRef]
- Salem, E.; Cook, E.A.J.; Lbacha, H.A.; Oliva, J.; Awoume, F.; Aplogan, G.L.; Hymann, E.C.; Muloi, D.; Deem, S.L.; Alali, S.; et al. Serologic evidence for influenza c and d virus among ruminants and camelids, africa, 1991-2015. Emerg Infect. Dis 2017, 23, 1556–1559. [Google Scholar] [CrossRef]
- Snoeck, C.J.; Oliva, J.; Pauly, M.; Losch, S.; Wildschutz, F.; Muller, C.P.; Hubschen, J.M.; Ducatez, M.F. Influenza d virus circulation in cattle and swine, luxembourg, 2012-2016. Emerg Infect. Dis 2018, 24, 1388–1389. [Google Scholar] [CrossRef]
- Zhai, S.L.; Zhang, H.; Chen, S.N.; Zhou, X.; Lin, T.; Liu, R.; Lv, D.H.; Wen, X.H.; Wei, W.K.; Wang, D.; et al. Influenza d virus in animal species in guangdong province, southern china. Emerg Infect. Dis 2017, 23, 1392–1396. [Google Scholar] [CrossRef]
- White, S.K.; Ma, W.; McDaniel, C.J.; Gray, G.C.; Lednicky, J.A. Serologic evidence of exposure to influenza d virus among persons with occupational contact with cattle. J. Clin. Virol 2016, 81, 31–33. [Google Scholar] [CrossRef]
- Sreenivasan, C.; Thomas, M.; Sheng, Z.; Hause, B.M.; Collin, E.A.; Knudsen, D.E.; Pillatzki, A.; Nelson, E.; Wang, D.; Kaushik, R.S.; et al. Replication and transmission of the novel bovine influenza d virus in a guinea pig model. J. Virol 2015, 89, 11990–12001. [Google Scholar] [CrossRef]
- Belser, J.A.; Eckert, A.M.; Tumpey, T.M.; Maines, T.R. Complexities in ferret influenza virus pathogenesis and transmission models. Microbiol Mol. Biol. Rev. 2016, 80, 733–744. [Google Scholar] [CrossRef]
- Rynda-Apple, A.; Robinson, K.M.; Alcorn, J.F. Influenza and bacterial superinfection: Illuminating the immunologic mechanisms of disease. Infect. Immun 2015, 83, 3764–3770. [Google Scholar] [CrossRef]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Metzger, D.W.; Sun, K. Immune dysfunction and bacterial coinfections following influenza. J. Immunol. 2013, 191, 2047–2052. [Google Scholar] [CrossRef]
- Robinson, K.M.; Kolls, J.K.; Alcorn, J.F. The immunology of influenza virus-associated bacterial pneumonia. Curr. Opin. Immunol. 2015, 34, 59–67. [Google Scholar] [CrossRef]
- Klonoski, J.M.; Watson, T.; Bickett, T.E.; Svendsen, J.M.; Gau, T.J.; Britt, A.; Nelson, J.T.; Schlenker, E.H.; Chaussee, M.S.; Rynda-Apple, A.; et al. Contributions of influenza virus hemagglutinin and host immune responses toward the severity of influenza virus: Streptococcus pyogenes superinfections. Viral Immunol. 2018, 31, 457–469. [Google Scholar] [CrossRef]
- Weeks-Gorospe, J.N.; Hurtig, H.R.; Iverson, A.R.; Schuneman, M.J.; Webby, R.J.; McCullers, J.A.; Huber, V.C. Naturally occurring swine influenza a virus pb1-f2 phenotypes that contribute to superinfection with gram-positive respiratory pathogens. J. Virol. 2012, 86, 9035–9043. [Google Scholar] [CrossRef]
- Shepardson, K.M.; Larson, K.; Johns, L.L.; Stanek, K.; Cho, H.; Wellham, J.; Henderson, H.; Rynda-Apple, A. Ifnar2 is required for anti-influenza immunity and alters susceptibility to post-influenza bacterial superinfections. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Shepardson, K.M.; Larson, K.; Morton, R.V.; Prigge, J.R.; Schmidt, E.E.; Huber, V.C.; Rynda-Apple, A. Differential type i interferon signaling is a master regulator of susceptibility to postinfluenza bacterial superinfection. MBio 2016, 7. [Google Scholar] [CrossRef]
- Shepardson, K.; Larson, K.; Cho, H.; Johns, L.L.; Malkoc, Z.; Stanek, K.; Wellhman, J.; Zaiser, S.; Daggs-Olson, J.; Moodie, T.; et al. A novel role for pdz-binding motif of influenza a virus nonstructural protein 1 in regulation of host susceptibility to postinfluenza bacterial superinfections. Viral Immunol. 2019, 32. [Google Scholar] [CrossRef]
- Lieber, M.; Smith, B.; Szakal, A.; Nelson-Rees, W.; Todaro, G. A continuous tumor-cell line from a human lung carcinoma with properties of type ii alveolar epithelial cells. Int. J. Cancer 1976, 17, 62–70. [Google Scholar] [CrossRef]
- Huber, V.C.; McKeon, R.M.; Brackin, M.N.; Miller, L.A.; Keating, R.; Brown, S.A.; Makarova, N.; Perez, D.R.; Macdonald, G.H.; McCullers, J.A. Distinct contributions of vaccine-induced immunoglobulin g1 (igg1) and igg2a antibodies to protective immunity against influenza. Clin. Vaccine Immunol. 2006, 13, 981–990. [Google Scholar] [CrossRef]
- Rynda-Apple, A.; Dobrinen, E.; McAlpine, M.; Read, A.; Harmsen, A.; Richert, L.E.; Calverley, M.; Pallister, K.; Voyich, J.; Wiley, J.A.; et al. Virus-like particle-induced protection against mrsa pneumonia is dependent on il-13 and enhancement of phagocyte function. Am. J. Pathol 2012, 181, 196–210. [Google Scholar] [CrossRef]
- Rynda-Apple, A.; Harmsen, A.; Erickson, A.S.; Larson, K.; Morton, R.V.; Richert, L.E.; Harmsen, A.G. Regulation of ifn-gamma by il-13 dictates susceptibility to secondary postinfluenza mrsa pneumonia. Eur. J. Immunol. 2014, 44, 3263–3272. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr. METHODS 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Groves, H.T.; McDonald, J.U.; Langat, P.; Kinnear, E.; Kellam, P.; McCauley, J.; Ellis, J.; Thompson, C.; Elderfield, R.; Parker, L.; et al. Mouse models of influenza infection with circulating strains to test seasonal vaccine efficacy. Front. Immunol 2018, 9, 126. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Lowen, A.C. Animal models for influenza virus pathogenesis and transmission. Viruses 2010, 2, 1530–1563. [Google Scholar] [CrossRef]
- Jia, L.; Xie, J.; Zhao, J.; Cao, D.; Liang, Y.; Hou, X.; Wang, L.; Li, Z. Mechanisms of severe mortality-associated bacterial co-infections following influenza virus infection. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef]
- Smith, A.M.; Adler, F.R.A.; Ribeiro, R.M.; Gutenkunst, R.N.; McAuley, J.L.; McCullers, J.A.; Perelson, A.S. Kinetics of coinfection with influenza a virus and streptococcus pneumoniae. PLoS Pathogens 2013, 9. [Google Scholar]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary bacterial infections associated with influenza pandemics. Front. Microbiol. 2017, 8, 1041. [Google Scholar] [CrossRef]
- Shirey, K.A.; Perkins, D.J.; Lai, W.; Zhang, W.; Fernando, L.R.; Gusovsky, F.; Blanco, J.C.G.; Vogel, S.N. Influenza “trains” the host for enhanced susceptibility to secondary bacterial infection. mBio 2019, 10. [Google Scholar] [CrossRef]
- Abramson, J.S.; Mills, E.L. Depression of neutrophil function induced by viruses and its role in secondary microbial infections. Rev. Infect. Dis 1988, 10, 326–341. [Google Scholar] [CrossRef]
- Shahangian, A.; Chow, E.K.; Tian, X.; Kang, J.R.; Ghaffari, A.; Liu, S.Y.; Belperio, J.A.; Cheng, G.; Deng, J.C. Type i ifns mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Investig. 2009, 119, 1910–1920. [Google Scholar] [CrossRef]
- Califano, D.; Furuya, Y.; Metzger, D.W. Effects of influenza on alveolar macrophage viability are dependent on mouse genetic strain. J. Immunol. 2018, 201, 134–144. [Google Scholar] [CrossRef]
- Jiang, H.; Shen, S.M.; Yin, J.; Zhang, P.P.; Shi, Y. Influenza virus nonstructural protein 1 inhibits the production of interferon beta of alveolar epithelial cells upon the infection of influenza a h1n1. Mol. Med. Rep. 2017, 16, 4553–4560. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and distinct functions of type i and type iii interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Sun, K.; Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 2008, 14, 558–564. [Google Scholar] [CrossRef]
- Robinson, K.M.; Lee, B.; Scheller, E.V.; Mandalapu, S.; Enelow, R.I.; Kolls, J.K.; Alcorn, J.F. The role of il-27 in susceptibility to post-influenza staphylococcus aureus pneumonia. Respir Res. 2015, 16, 10. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skelton, R.M.; Shepardson, K.M.; Hatton, A.; Wilson, P.T.; Sreenivasan, C.; Yu, J.; Wang, D.; Huber, V.C.; Rynda-Apple, A. RETRACTED: Contribution of Host Immune Responses against Influenza D Virus Infection toward Secondary Bacterial Infection in a Mouse Model. Viruses 2019, 11, 994. https://doi.org/10.3390/v11110994
Skelton RM, Shepardson KM, Hatton A, Wilson PT, Sreenivasan C, Yu J, Wang D, Huber VC, Rynda-Apple A. RETRACTED: Contribution of Host Immune Responses against Influenza D Virus Infection toward Secondary Bacterial Infection in a Mouse Model. Viruses. 2019; 11(11):994. https://doi.org/10.3390/v11110994
Chicago/Turabian StyleSkelton, Raegan M., Kelly M. Shepardson, Alexis Hatton, Patrick T. Wilson, Chithra Sreenivasan, Jieshi Yu, Dan Wang, Victor C. Huber, and Agnieszka Rynda-Apple. 2019. "RETRACTED: Contribution of Host Immune Responses against Influenza D Virus Infection toward Secondary Bacterial Infection in a Mouse Model" Viruses 11, no. 11: 994. https://doi.org/10.3390/v11110994
APA StyleSkelton, R. M., Shepardson, K. M., Hatton, A., Wilson, P. T., Sreenivasan, C., Yu, J., Wang, D., Huber, V. C., & Rynda-Apple, A. (2019). RETRACTED: Contribution of Host Immune Responses against Influenza D Virus Infection toward Secondary Bacterial Infection in a Mouse Model. Viruses, 11(11), 994. https://doi.org/10.3390/v11110994