Phylodynamic and Phylogeographic Analysis of Hepatitis Delta Virus Genotype 3 Isolated in South America

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Alignment and Determination of Evolutionary Model
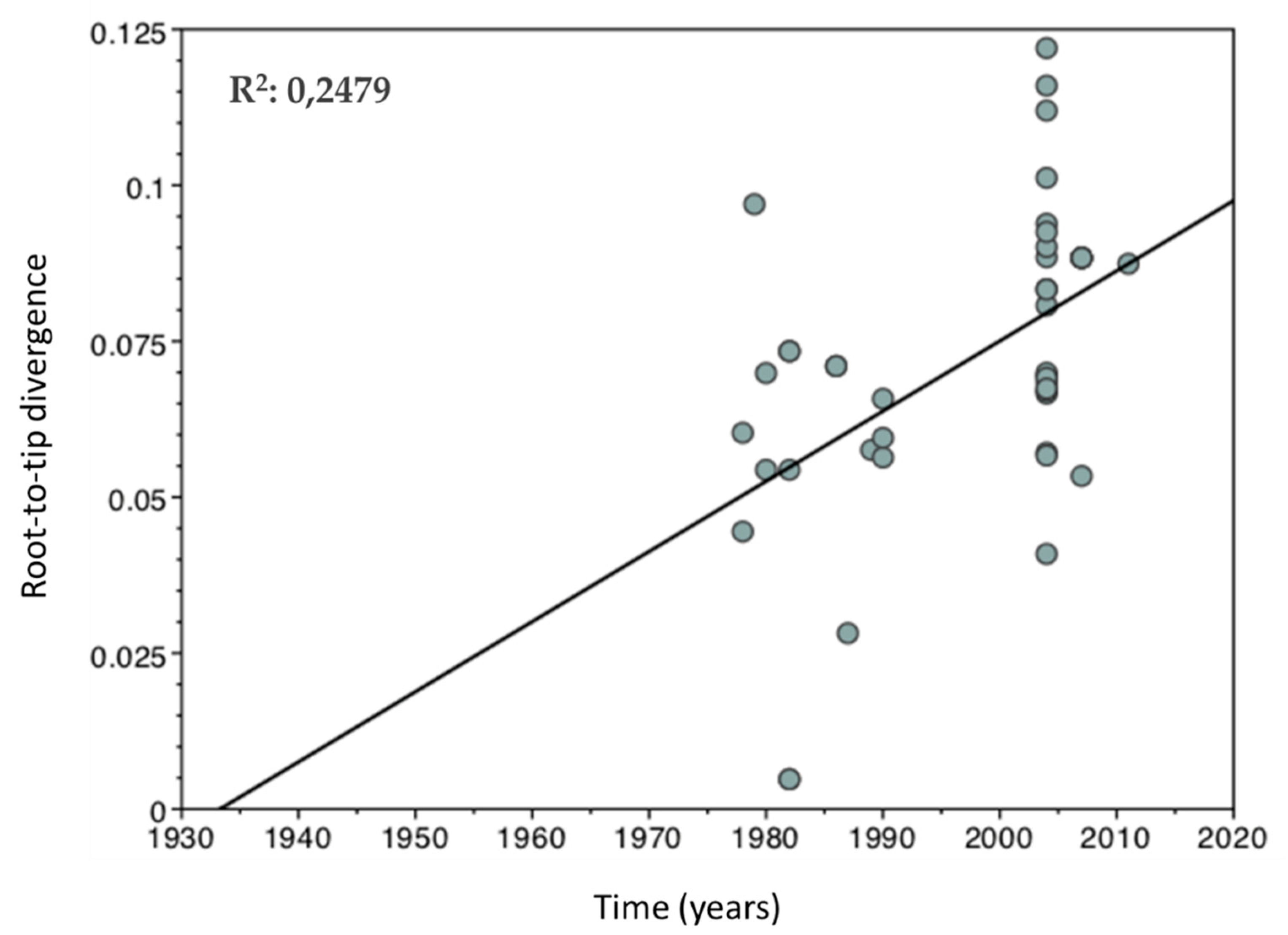
2.3. Temporal Signal Estimation
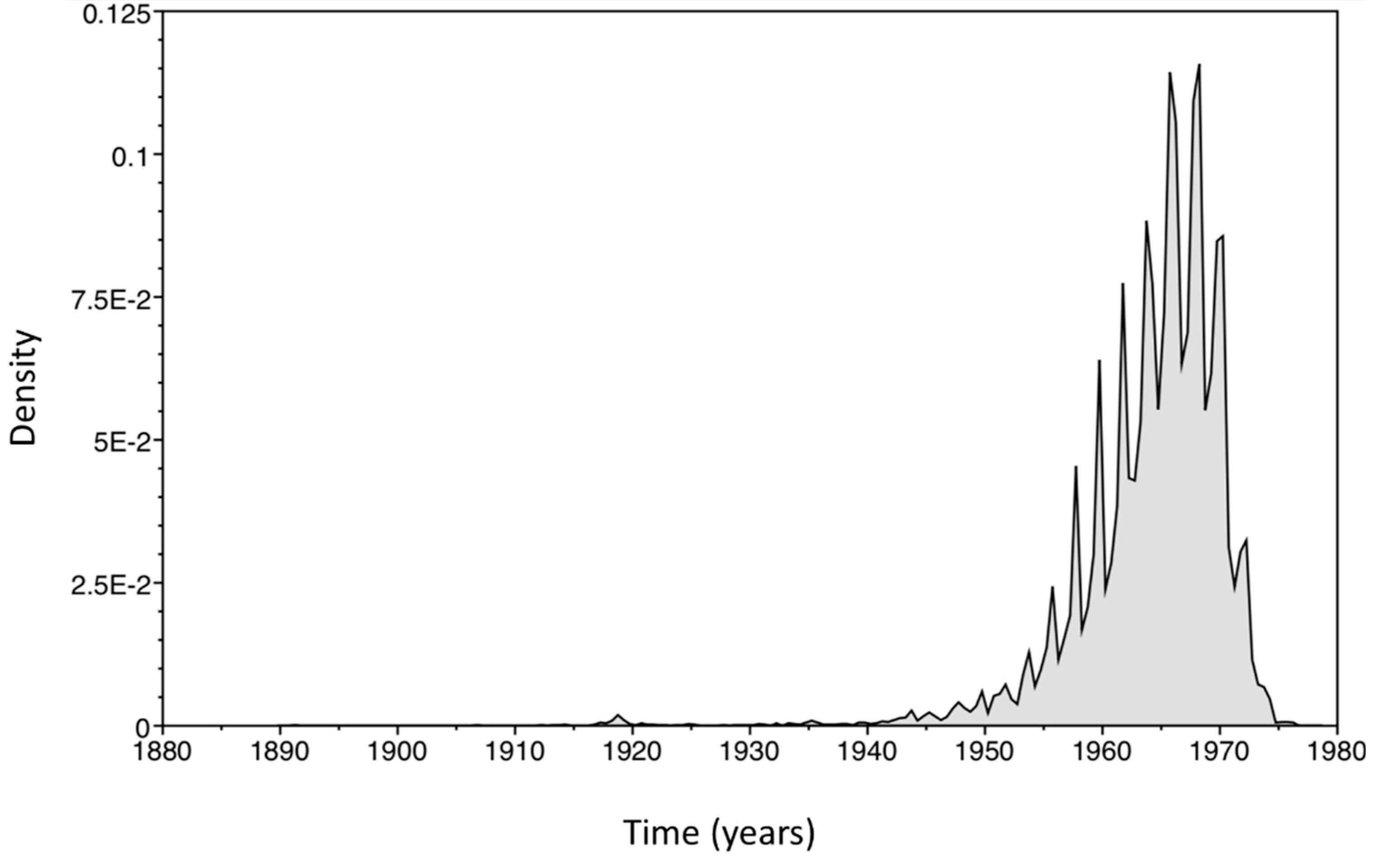
2.4. Bayesian Analysis
2.5. Spatial Phylogenetic Reconstruction of the Evolutionary Dynamics
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cunha, C.; Tavanez, J.P.; Gudima, S. Hepatitis delta virus: A fascinating and neglected pathogen. World J. Virol. 2015, 4, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Facchetti, F.; Perbellini, R.; Galmozzi, E.; Petruzziello, A.; Di Capua, L.; Sabatino, R.; Botti, G.; Loquercio, G.; Pecheur, E.I.; et al. Hepatitis delta virus and hepatocellular carcinoma: An update. Epidemiol. Infect. 2018, 146, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Dandri, M. Hepatitis B and Delta Virus: Advances on Studies about Interactions between the Two Viruses and the Infected Hepatocyte. J. Clin. Transl. Hepatol. 2015, 3, 220–229. [Google Scholar]
- Botelho-Souza, L.F.; Vasconcelos, M.P.A.; dos Santos, A.O.; Salcedo, J.M.V.; Vieira, D.S. Hepatitis delta: Virological and clinical aspects. Virol. J. 2017, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Urban, S. Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen. Viruses 2017, 9, 172. [Google Scholar] [CrossRef]
- Taylor, J.M. Host RNA circles and the origin of hepatitis delta virus. World J. Gastroenterol. 2014, 20, 2971. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, S.; Ma, Z.; Hakim, M.S.; Wang, W.; Peppelenbosch, M.P.; Pan, Q. Recombinant identification, molecular classification and proposed reference genomes for hepatitis delta virus. J. Viral Hepat. 2019, 26, 183–190. [Google Scholar] [CrossRef]
- Le Gal, F.; Brichler, S.; Drugan, T.; Alloui, C.; Roulot, D.; Pawlotsky, J.M.; Dény, P.; Gordien, E. Genetic diversity and worldwide distribution of the deltavirus genus: A study of 2,152 clinical strains. Hepatology 2017, 66, 1826–1841. [Google Scholar] [CrossRef]
- Rizzetto, M.; Ponzetto, A.; Forzani, I. Hepatitis delta virus as a global health problem. Vaccine 1990, 8, S10–S14, discussion S21–S23. [Google Scholar] [CrossRef]
- Hughes, S.A.; Wedemeyer, H.; Harrison, P.M. Hepatitis delta virus. Lancet 2011, 378, 73–85. [Google Scholar] [CrossRef]
- Barros, L.M.F.; Gomes-Gouvêa, M.S.; Pinho, J.R.R.; Alvarado-Mora, M.V.; Dos Santos, A.; Mendes-Corrêa, M.C.J.; Caldas, A.J.M.; Sousa, M.T.; Santos, M.D.C.; Ferreira, A.S.P. Hepatitis Delta virus genotype 8 infection in Northeast Brazil: Inheritance from African slaves? Virus Res. 2011, 160, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.D.C.; Gomes-Gouvêa, M.S.; Nunes, J.D.C.; Barros, L.M.F.; Carrilho, F.J.; Ferreira, A.D.S.P.; Pinho, J.R.R. The hepatitis delta genotype 8 in Northeast Brazil: The North Atlantic slave trade as the potential route for infection. Virus Res. 2016, 224, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Brasil, Ministério da Saúde. Boletim epidemiológico das hepatites virais. Secretaria de Vigilância em Saúde, Ministério da Saúde 2018, 49, 6–67. [Google Scholar]
- Gomes-Gouvêa, M.S.; Soares, M.D.C.P.; de Carvalho Mello, I.M.V.G.; Brito, E.M.F.; Moia, L.D.J.M.P.; Bensabath, G.; Nunes, H.M.; Carrilho, F.J.; Pinho, J.R.R. Hepatitis D and B virus genotypes in chronically infected patients from the Eastern Amazon Basin. Acta Trop. 2008, 106, 149–155. [Google Scholar] [CrossRef]
- Botelho-Souza, L.F.; Souza Vieira, D.; de Oliveira dos Santos, A.; Cunha Pereira, A.V.; Villalobos-Salcedo, J.M. Characterization of the Genotypic Profile of Hepatitis Delta Virus: Isolation of HDV Genotype-1 in the Western Amazon Region of Brazil. Intervirology 2015, 58, 166–171. [Google Scholar] [CrossRef]
- Di Filippo Villa, D.; Cortes-Mancera, F.; Payares, E.; Montes, N.; de la Hoz, F.; Arbelaez, M.P.; Correa, G.; Navas, M.C. Hepatitis D virus and hepatitis B virus infection in Amerindian communities of the Amazonas state, Colombia. Virol. J. 2015, 12, 172. [Google Scholar] [CrossRef]
- Cicero, M.F.; Pena, N.M.; Santana, L.C.; Arnold, R.; Azevedo, R.G.; Leal, É.D.S.; Diaz, R.S.; Komninakis, S.V. Is Hepatitis Delta infections important in Brazil? BMC Infect. Dis. 2016, 16, 525. [Google Scholar] [CrossRef]
- Botelho Souza, L.F. Análise Genomica do Vírus da Hepatite Delta Isolado na Amazônia Ocidental. Tese (Doutorado em Biologia Experimental) - Programa de Pós-graduação em Biologia experimental da Fundação; Universidade Federal de Rondônia/UNIR: Porto Velho, Brazil, 2018. [Google Scholar]
- Casey, J.L.; Brown, T.L.; Colan, E.J.; Wignall, F.S.; Gerin, J.L. A genotype of hepatitis D virus that occurs in northern South America. Proc. Natl. Acad. Sci. USA. 1993, 90, 9016–9020. [Google Scholar] [CrossRef]
- Viana, S.; Paraná, R.; Moreira, R.C.; Compri, A.P.; Macedo, V. High prevalence of hepatitis B virus and hepatitis D virus in the western Brazilian Amazon. Am. J. Trop. Med. Hyg. 2005, 73, 808–814. [Google Scholar] [CrossRef]
- Paraná, R.; Kay, A.; Molinet, F.; Viana, S.; Silva, L.K.; Salcedo, J.M.; Tavares-Neto, J.; Lobato, C.; Rios-Leite, M.; Matteoni, L.; et al. HDV genotypes in the Western Brazilian Amazon region: A preliminary report. Am. J. Trop. Med. Hyg. 2006, 75, 475–479. [Google Scholar] [CrossRef]
- Gomes-Gouvea, M.S.; Soares, M.C.P.; Bensabath, G.; de Carvalho-Mello, I.M.V.G.; Brito, E.M.F.; Souza, O.S.C.; Queiroz, A.T.L.; Carrilho, F.J.; Pinho, J.R.R. Hepatitis B virus and hepatitis delta virus genotypes in outbreaks of fulminant hepatitis (Labrea black fever) in the western Brazilian Amazon region. J. Gen. Virol. 2009, 90, 2638–2643. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Canese, M.G.; Aricò, S.; Crivelli, O.; Trepo, C.; Bonino, F.; Verme, G. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut 1977, 18, 997. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Mora, M.V.; Romano, C.M.; Gomes-Gouvêa, M.S.; Gutierrez, M.F.; Carrilho, F.J.; Pinho, J.R.R. Dynamics of Hepatitis D (delta) virus genotype 3 in the Amazon region of South America. Infect. Genet. Evol. 2011, 11, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Azarbahra, M.; Tajbakhsh, E.; Momtaz, H. Phylogenetic analysis of hepatitis delta virus isolated from HBsAg positive patients in Shahrekord, Iran. Asian Pacific. J. Trop. Dis. 2014, 4, 391–397. [Google Scholar] [CrossRef]
- Wu, J.C.; Chiang, T.Y.; Sheen, I.J. Characterization and phylogenetic analysis of a novel hepatitis D virus strain discovered by restriction fragment length polymorphism analysis. J. Gen. Virol. 1998, 79, 1105–1113. [Google Scholar] [CrossRef][Green Version]
- Crispim, M.A.E.; Fraiji, N.A.; Campello, S.C.; Schriefer, N.A.; Stefani, M.M.A.; Kiesslich, D. Molecular epidemiology of hepatitis B and hepatitis delta viruses circulating in the Western Amazon region, North Brazil. BMC Infect. Dis. 2014, 14, 94. [Google Scholar] [CrossRef]
- Nakano, T.; Hadler, S.C.; Orito, E.; Shapiro, C.N.; Casey, J.L.; Mizokami, M.; Robertson, B.H. Characterization of hepatitis D virus genotype III among Yucpa Indians in Venezuela. J. Gen. Virol. 2001, 82, 2183–2189. [Google Scholar] [CrossRef]
- Marques, V.A.; Lewis-Ximenez, L.L.S.R.; Lampe, E. Complete Genome Sequence of Hepatitis D Virus Isolated in Brazil. Available online: https://www.ncbi.nlm.nih.gov/nuccore/kc590319.1 (accessed on 16 July 2019).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Nogueira-Lima, F.S. Oswaldo Cruz Foundation of Rondônia, Porto Velho, Rondônia. Complementary analysis with 55 HDV-1 sequences also obtained from Nucleotide database to compare temporal signal quality between different sequence sizes analyzed: Complete genome and R0 region. 2019. [Google Scholar]
- Hadler, S.C.; De Monzon, M.A.; Rivero, D.; Perez, M.; Bracho, A.; Fields, H. Epidemiology and long-term consequences of hepatitis delta virus infection in the yucpa Indians of venezuela. Am. J. Epidemiol. 1992, 136, 1507–1516. [Google Scholar] [CrossRef]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, K.E.; Patarroyo, M.E.; Engle, R.; Purcell, R.H.; Gerin, J.L. Viral hepatitis in Colombia: A study of the hepatitis of the Sierra Nevada de Santa Marta. Hepatology 1985, 5, 299–304. [Google Scholar] [PubMed]
- Buitrago, B.; Popper, H.; Hadler, S.C.; Thung, S.N.; Gerber, M.A.; Purcell, R.H.; Maynard, J.E. Specific histologic features of Santa Marta hepatitis: A severe form of hepatitis delta-virus infection in northern South America. Hepatology 1986, 6, 1285–1291. [Google Scholar] [CrossRef]
- Bensabath, G. Presença do Agente Delta associado-VHB em residentes do Município de Boca do Acre, micro região do Purus, Amazonas. In Proceedings of the Nota Prévia. Programa E Resumos, XIX Congresso da Sociedade Brasileira de Medicina Tropical, Rio de Janeiro, Brazil; 1983; pp. 150–151. [Google Scholar]
- Dias, L.B.; Coura, J.R. Hepatite de Lábrea: Estudo de revisão em viscerotomias hepáticas dos anos de 1934 a 1940. Rev. Inst. Med. Trop. Sao Paulo 1985, 27, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.C.; Ferreira, L.C.; Brasil, L.M.; Castilho, M.D.C.; Moss, R.; Barone, M. Fulminant Labrea hepatitis—The role of hepatitis A (HAV), B (HBV), C (HCV), and D (HDV) infection. (Preliminary report). Rev. Inst. Med. Trop. Sao Paulo 1992, 34, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.C.F. da Histórico das hepatites virais. Rev. Soc. Bras. Med. Trop. 2010, 43, 322–330. [Google Scholar] [CrossRef]
- Braga, W.S.M.; Melo, H.O.; Cossate, M.D.B.; Castilho, M.C.; Souza, R.A.B.; Brasil, L.M.; Fonseca, J.C.F. Prevalência dos marcadores sorológicos dos vírus da hepatite B e Delta em população assintomática: Estudo do impacto do uso da vacina contra hepatite B em áreas hiperendêmicas, Itamarati-Amazonas, Vale do rio Juruá. Rev. da Soc. Bras. Med. Trop. 1998, 31, 31. [Google Scholar]
- Castilho, M.C. Aspectos Epidemiológicos e da Biologia Molecular da Hepatite B em três Comunidades da Amazônia Ocidental Brasileira. Ph.D. Thesis, Medicina Tropical da Universidade do Estado do Amazonas, Manaus, Brazil, 2012. [Google Scholar]
- Scarponi, C.F.D.O.; Silva, R.D.N.D.; Souza Filho, J.A.D.; Guerra, M.R.L.; Pedrosa, M.A.F.; Mol, M.P.G. Hepatitis Delta Prevalence in South America: A Systematic Review and Meta-Analysis. Rev. Soc. Bras. Med. Trop. 2019, 52. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Access number | Country of Isolation | Collection date | Clinical information | Author |
|---|---|---|---|---|
| * KF278974.1 to KF278994.1 | Brazil | Sep/2004 | CH | [27] |
| * EU287868.1 to EU287872.1 | Colombia | Nov/2007 | FH | [24] |
| * AB037947.1 to AB037949.1 | Venezuela | 1990 | FH | [28] |
| FJ010634.1 | Brazil | Nov/1986 | FH+D | [22] |
| FJ010635.1 | Brazil | Nov/1986 | FH+D | [22] |
| FJ010636.1 | Brazil | May/1982 | FH+D | [22] |
| FJ010637.1 | Brazil | Nov/1980 | FH+D | [22] |
| FJ010638.1 | Brazil | Jul/1989 | FH+D | [22] |
| FJ010639.1 | Brazil | Sep/1982 | FH+D | [22] |
| FJ010640.1 | Brazil | Nov/1982 | FH+D | [22] |
| FJ010641.1 | Brazil | Sep/1987 | FH+D | [22] |
| FJ010642.1 | Brazil | Aug/1978 | FH+D | [22] |
| FJ010643.1 | Brazil | Sep/1978 | FH+D | [22] |
| FJ010644.1 | Brazil | Jun/1982 | FH+D | [22] |
| FJ010645.1 | Brazil | May/1982 | FH+D | [22] |
| FJ010646.1 | Brazil | Sep/1979 | FH+D | [22] |
| FJ010647.1 | Brazil | Nov/1980 | FH+D | [22] |
| KC590319.1 | Brazil | Jun/2011 | NA | [29] |
| Sequence | Latitude | Longitude | Place of isolation |
|---|---|---|---|
| KF278994.1_HDV-3_Brazil_Sep/2004 | −3.368333 | −64.719167 | Tefé (Amazonas, Brazil) |
| KF278993.1_HDV-3_Brazil_Sep/2004 | −7.260000 | −64.799167 | Labrea (Amazonas, Brazil) |
| KF278992.1_HDV-3_Brazil_Sep/2004 | −0.130278 | −67.089167 | S.Gabriel da Cachoeira (Amazonas, Brazil) |
| KF278991.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil) |
| KF278990.1_HDV-3_Brazil_Sep/2004 | −5.628333 | −63.183611 | Tapaua (Amazonas, Brazil) |
| KF278989.1_HDV-3_Brazil_Sep/2004 | −3.368333 | −64.719167 | Tefe (Amazonas, Brazil) |
| KF278988.1_HDV-3_Brazil_Sep/2004 | −7.260000 | −64.799167 | Labrea (Amazonas, Brazil) |
| KF278987.1_HDV-3_Brazil_Sep/2004 | −3.837222 | −62.057500 | Codajas (Amazonas, Brazil) |
| KF278986.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil) |
| KF278985.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil |
| KF278984.1_HDV-3_Brazil_Sep/2004 | −7.260000 | −64.799167 | Labrea (Amazonas, Brazil) |
| KF278983.1_HDV-3_Brazil_Sep/2004 | −7.260000 | −64.799167 | Labrea (Amazonas, Brazil) |
| KF278982.1_HDV-3_Brazil_Sep/2004 | −7.714444 | −66.976389 | Pauini (Amazonas, Brazil) |
| KF278981.1_HDV-3_Brazil_Sep/2004 | −7.051389 | −71.695556 | Ipixuna (Amazonas, Brazil) |
| KF278980.1_HDV-3_Brazil_Sep/2004 | −3.812222 | −60.345556 | Careiro (Amazonas, Brazil) |
| KF278979.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil) |
| KF278978.1_HDV-3_Brazil_Sep/2004 | −8.135556 | −70.765000 | Tarauaca (Acre, Brazil) |
| KF278977.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil) |
| KF278976.1_HDV-3_Brazil_Sep/2004 | −6.533889 | −64.383056 | Canutama (Amazonas, Brazil) |
| KF278975.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil) |
| KF278974.1_HDV-3_Brazil_Sep/2004 | −6.660278 | −69.874444 | Eirunepe (Amazonas, Brazil) |
| EU287872.1_HDV-3_Colombia_Nov/2007 | −1.416944 | −71.577778 | Amazonas/Colombia |
| EU287871.1_HDV-3_Colombia_Nov/2007 | −1.416944 | −71.577778 | Amazonas/Colombia |
| EU287869.1_HDV-3_Colombia_Nov/2007 | −1.416944 | −71.577778 | Amazonas/Colombia |
| EU287870.1_HDV-3_Colombia_Nov/2007 | −1.416944 | −71.577778 | Amazonas/Colombia |
| EU287868.1_HDV-3_Colombia_Nov/2007 | −1.416944 | −71.577778 | Amazonas/Colombia |
| FJ010647.1_HDV-3_Brazil_22-Nov-1980 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010646.1_HDV-3_Brazil_Sep/1979 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010645.1_HDV-3_Brazil_May/1982 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010644.1_HDV-3_Brazil_Jun/1982 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010643.1_HDV-3_Brazil_Sep/1978 | −906721 | −68.6577 | Sena Madueira (Acre, Brazil) |
| FJ010642.1_HDV-3_Brazil_Aug/1978 | −906721 | −68.6577 | Sena Madueira (Acre, Brazil) |
| FJ010641.1_HDV-3_Brazil_Sep/1987 | −7.714444 | −66.976389 | Pauini (Amazonas, Brazil) |
| FJ010640.1_HDV-3_Brazil_Nov/1982 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010639.1_HDV-3_Brazil_Sep/1982 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010638.1_HDV-3_Brazil_Jul/1989 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010637.1_HDV-3_Brazil_Nov/1980 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010636.1_HDV-3_Brazil_May/1982 | −8.740556 | −67.384167 | Boca do Acre (Amazonas, Brazil) |
| FJ010635.1_HDV-3_Brazil_Aug/1986 | −7.714444 | −66.976389 | Pauini (Amazonas, Brazil) |
| FJ010634.1_HDV-3_Brazil_Nov/1986 | −7.714444 | −66.976389 | Pauini (Amazonas, Brazil) |
| KC590319.1_HDV-3_Brazil_Jun/2011 | −3.750000 | −64.500000 | Amazonas/Brazil |
| AB037948.1_HDV-3_Venezuela_1990 | 10.038056 | −73.011944 | Sierra De Perija (Zulia, Venezuela) |
| AB037947.1_HDV-3_Venezuela_1990 | 10.038056 | −73.011944 | Sierra De Perija (Zulia, Venezuela) |
| AB037949.1_HDV-3_Venezuela_1990 | 10.038056 | −73.011944 | Sierra De Perija (Zulia, Venezuela) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira-Lima, F.S.; Botelho-Souza, L.F.; Roca, T.P.; Santos, A.O.d.; Oliveira, S.d.C.; Queiroz, J.A.d.S.; Santos-Alves, F.A.G.d.; Salcedo, J.M.V.; Vieira, D.S. Phylodynamic and Phylogeographic Analysis of Hepatitis Delta Virus Genotype 3 Isolated in South America. Viruses 2019, 11, 995. https://doi.org/10.3390/v11110995
Nogueira-Lima FS, Botelho-Souza LF, Roca TP, Santos AOd, Oliveira SdC, Queiroz JAdS, Santos-Alves FAGd, Salcedo JMV, Vieira DS. Phylodynamic and Phylogeographic Analysis of Hepatitis Delta Virus Genotype 3 Isolated in South America. Viruses. 2019; 11(11):995. https://doi.org/10.3390/v11110995
Chicago/Turabian StyleNogueira-Lima, Felipe Souza, Luan Felipo Botelho-Souza, Tárcio Peixoto Roca, Alcione Oliveira dos Santos, Suyane da Costa Oliveira, Jackson Alves da Silva Queiroz, Fabianne Araújo Gomes dos Santos-Alves, Juan Miguel Villalobos Salcedo, and Deusilene Souza Vieira. 2019. "Phylodynamic and Phylogeographic Analysis of Hepatitis Delta Virus Genotype 3 Isolated in South America" Viruses 11, no. 11: 995. https://doi.org/10.3390/v11110995
APA StyleNogueira-Lima, F. S., Botelho-Souza, L. F., Roca, T. P., Santos, A. O. d., Oliveira, S. d. C., Queiroz, J. A. d. S., Santos-Alves, F. A. G. d., Salcedo, J. M. V., & Vieira, D. S. (2019). Phylodynamic and Phylogeographic Analysis of Hepatitis Delta Virus Genotype 3 Isolated in South America. Viruses, 11(11), 995. https://doi.org/10.3390/v11110995