Towards Understanding KSHV Fusion and Entry


Abstract
:1. Introduction
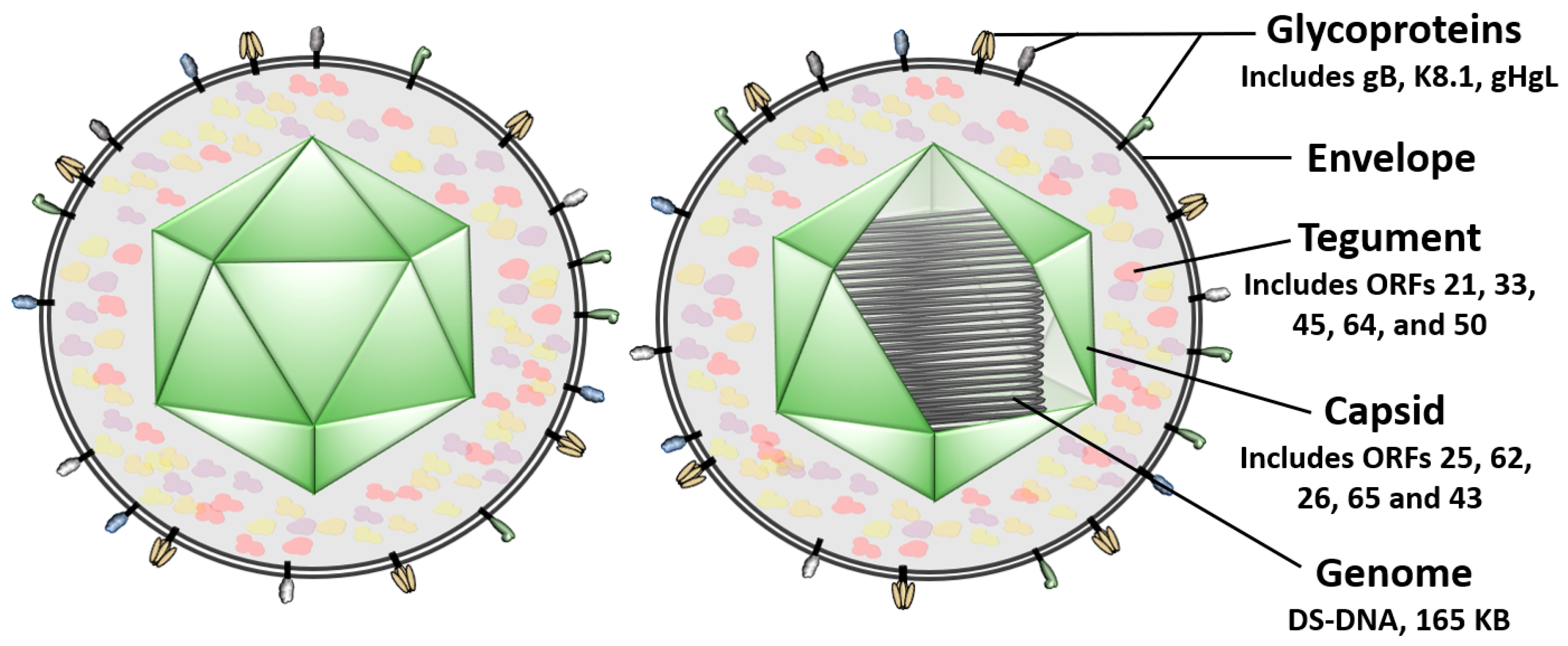
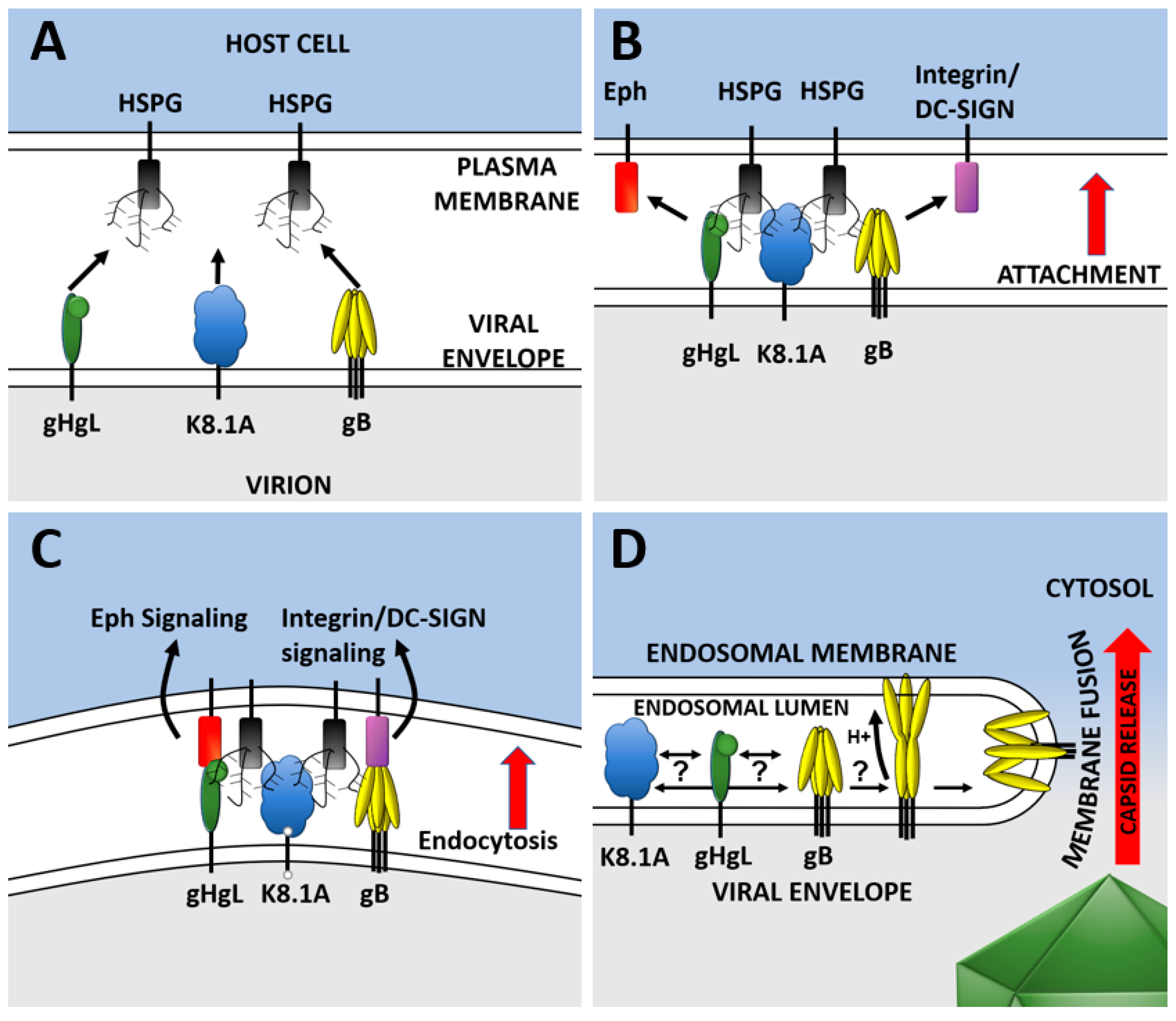
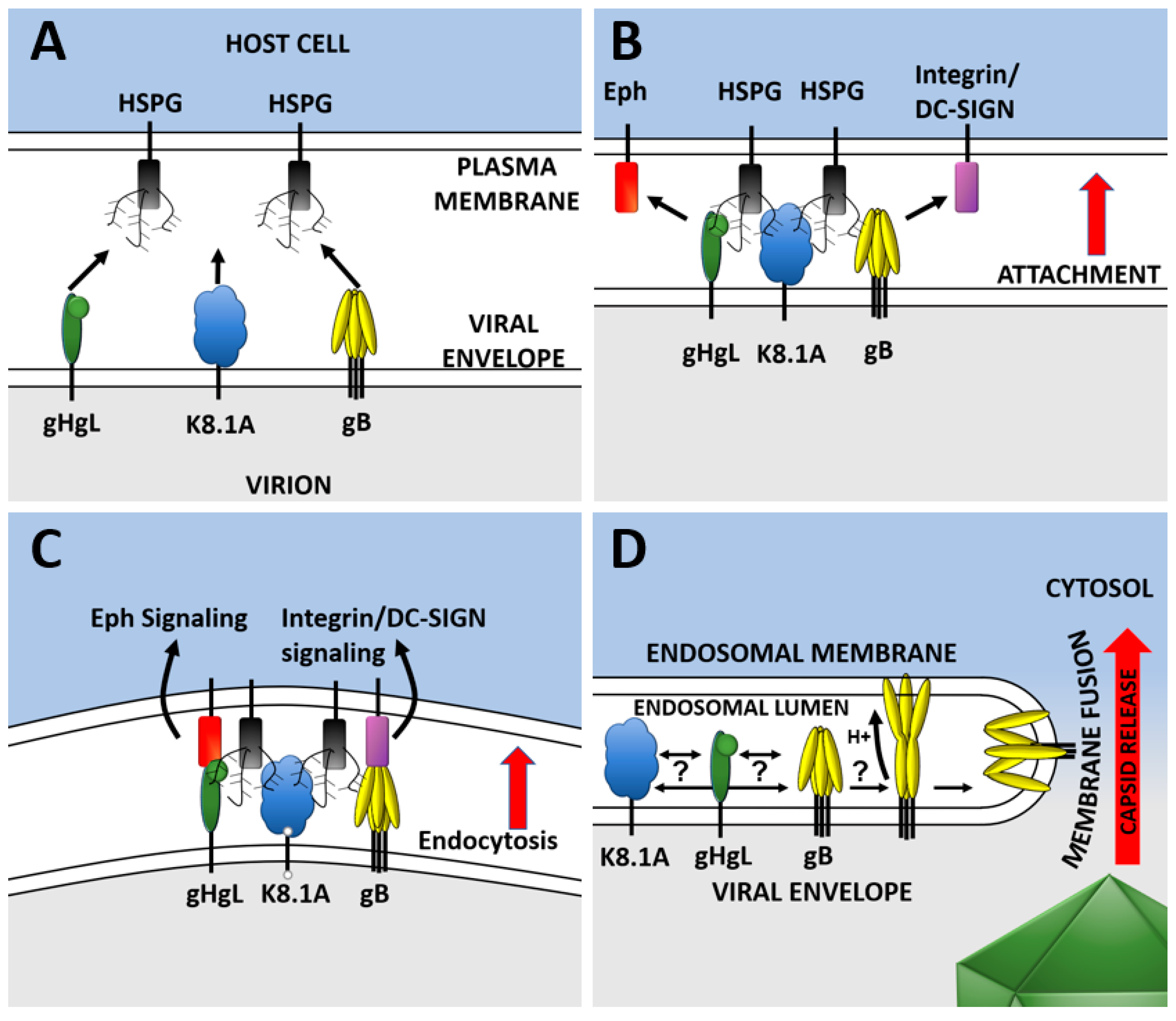
2. KSHV Entry
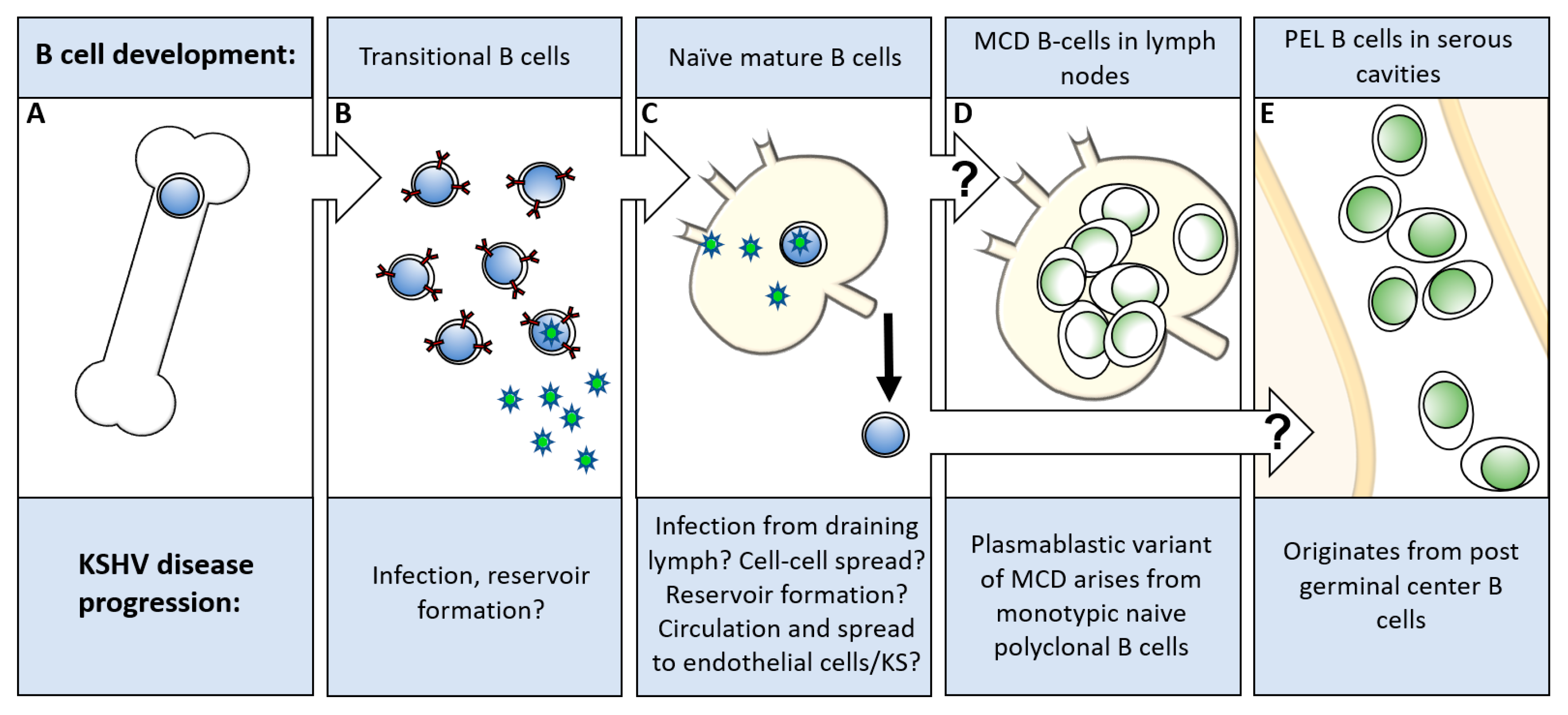
3. Host Cell Tropism
4. KSHV Entry Glycoproteins
4.1. K8.1
4.2. Glycoprotein B
4.3. Glycoproteins H and L
4.4. Other Virion Glycoproteins
4.5. Cell Associated Virus/Cell-to-Cell Spread
4.6. Fusion
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Moore, P.S.; Gao, S.J.; Dominguez, G.; Cesarman, E.; Lungu, O.; Knowles, D.M.; Garber, R.; Pellett, P.E.; McGeoch, D.J.; Chang, Y. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J. Virol. 1996, 70, 549–558. [Google Scholar] [PubMed]
- ICTV. Virus Taxonomy: 2018b Release; International Committee on Taxonomy of Viruses: Washington, DC, USA, 2018. [Google Scholar]
- Roizman, B.; Carmichael, L.E.; Deinhardt, F.; de-The, G.; Nahmias, A.J.; Plowright, W.; Rapp, F.; Sheldrick, P.; Takahashi, M.; Wolf, K. Herpesviridae. Definition, provisional nomenclature, and taxonomy. The Herpesvirus Study Group, the International Committee on Taxonomy of Viruses. InterVirology 1981, 16, 201–217. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Kaposi, M. Idiopathisches multiples Pigmentsarkomen der. Haut. Archiv Dermatol Syph. 1872, 1872, 265–273. [Google Scholar] [CrossRef]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. Off. Pub. Infect. Dis. Soc. Am. 2010, 51, 350–358. [Google Scholar] [CrossRef]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical Manifestations of Kaposi Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front. Microbiol. 2012, 3, 73. [Google Scholar] [CrossRef]
- Henke-Gendo, C.; Schulz, T.F. Transmission and disease association of Kaposi’s sarcoma-associated herpesvirus: Recent developments. Curr. Opin. Infect. Dis. 2004, 17, 53–57. [Google Scholar] [CrossRef]
- Pica, F.; Volpi, A. Transmission of human herpesvirus 8: An update. Curr. Opin. Infect. Dis. 2007, 20, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Dedicoat, M.; Newton, R.; Alkharsah, K.R.; Sheldon, J.; Szabados, I.; Ndlovu, B.; Page, T.; Casabonne, D.; Gilks, C.F.; Cassol, S.A.; et al. Mother-to-child transmission of human herpesvirus-8 in South Africa. J. Infect. Dis. 2004, 190, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Labo, N.; Wakeham, K.; Marshall, V.; Roshan, R.; Nalwoga, A.; Sebina, I.; Muhangi, L.; Webb, E.L.; Miley, W.; et al. Determinants of Gammaherpesvirus Shedding in Saliva Among Ugandan Children and Their Mothers. J. Infect. Dis. 2018, 218, 892–900. [Google Scholar] [CrossRef]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Lagunoff, M.; Zhong, W.; Ganem, D. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 1996, 70, 8151–8154. [Google Scholar] [PubMed]
- Dai, X.; Gong, D.; Wu, T.T.; Sun, R.; Zhou, Z.H. Organization of capsid-associated tegument components in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 12694–12702. [Google Scholar] [CrossRef]
- Bechtel, J.; Grundhoff, A.; Ganem, D. RNAs in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 10138–10146. [Google Scholar] [CrossRef]
- Bechtel, J.T.; Winant, R.C.; Ganem, D. Host and viral proteins in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 4952–4964. [Google Scholar] [CrossRef]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef]
- Rozen, R.; Sathish, N.; Li, Y.; Yuan, Y. Virion-wide protein interactions of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 4742–4750. [Google Scholar] [CrossRef]
- Said, J.W.; Chien, K.; Tasaka, T.; Koeffler, H.P. Ultrastructural characterization of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) in Kaposi’s sarcoma lesions: Electron microscopy permits distinction from cytomegalovirus (CMV). J. Pathol. 1997, 182, 273–281. [Google Scholar] [CrossRef]
- Mohl, B.S.; Chen, J.; Longnecker, R. Gammaherpesvirus entry and fusion: A tale how two human pathogenic viruses enter their host cells. Adv. Virus Res. 2019, 104, 313–343. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interaction of KSHV with host cell surface receptors and cell entry. Viruses 2014, 6, 4024–4046. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Dai, X.; Xiao, Y.; Du, Y.; Chapa, T.J.; Johnson, J.R.; Li, X.; Krogan, N.J.; Deng, H.; Wu, T.T.; et al. Virus-Like Vesicles of Kaposi’s Sarcoma-Associated Herpesvirus Activate Lytic Replication by Triggering Differentiation Signaling. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Zhu, F.X.; Yuan, Y. The ORF45 protein of Kaposi’s sarcoma-associated herpesvirus is associated with purified virions. J. Virol. 2003, 77, 4221–4230. [Google Scholar] [CrossRef]
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Cell-homologous genes in the Kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: Determinants of its pathogenicity? J. Virol. 1997, 71, 4187–4192. [Google Scholar]
- Naranatt, P.P.; Akula, S.M.; Chandran, B. Characterization of gamma2-human herpesvirus-8 glycoproteins gH and gL. Arch. Virol. 2002, 147, 1349–1370. [Google Scholar] [CrossRef]
- Baghian, A.; Luftig, M.; Black, J.B.; Meng, Y.X.; Pau, C.P.; Voss, T.; Pellett, P.E.; Kousoulas, K.G. Glycoprotein B of human herpesvirus 8 is a component of the virion in a cleaved form composed of amino-and carboxyl-terminal fragments. Virology 2000, 269, 18–25. [Google Scholar] [CrossRef]
- Akula, S.M.; Wang, F.Z.; Vieira, J.; Chandran, B. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 2001, 282, 245–255. [Google Scholar] [CrossRef]
- Koyano, S.; Mar, E.C.; Stamey, F.R.; Inoue, N. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J. Gen. Virol. 2003, 84, 1485–1491. [Google Scholar] [CrossRef]
- Birkmann, A.; Mahr, K.; Ensser, A.; Yaguboglu, S.; Titgemeyer, F.; Fleckenstein, B.; Neipel, F. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J. Virol. 2001, 75, 11583–11593. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 2001, 284, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Dollery, S.J.; Santiago-Crespo, R.J.; Chatterjee, D.; Berger, E.A. Glycoprotein K8.1A of Kaposi’s Sarcoma-Associated Herpesvirus Is a Critical B Cell Tropism Determinant Independent of Its Heparan Sulfate Binding Activity. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, L.; Lee, W.H.; Spiller, O.B.; Villoutreix, B.O.; Blom, A.M. The Kaposi’s sarcoma-associated herpesvirus complement control protein (KCP) binds to heparin and cell surfaces via positively charged amino acids in CCP1-2. Mol. Immunol. 2006, 43, 1665–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniraju, M.; Mutsvunguma, L.Z.; Foley, J.; Escalante, G.M.; Rodriguez, E.; Nabiee, R.; Totonchy, J.; Mulama, D.H.; Nyagol, J.; Wussow, F.; et al. Kaposi Sarcoma-Associated Herpesvirus Glycoprotein H Is Indispensable for Infection of Epithelial, Endothelial, and Fibroblast Cell Types. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TerBush, A.A.; Hafkamp, F.; Lee, H.J.; Coscoy, L. A Kaposi’s Sarcoma-Associated Herpesvirus Infection Mechanism Is Independent of Integrins alpha3beta1, alphaVbeta3, and alphaVbeta5. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Veettil, M.V.; Sadagopan, S.; Sharma-Walia, N.; Wang, F.Z.; Raghu, H.; Varga, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J. Virol. 2008, 82, 12126–12144. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; Konig, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.S.; Desrosiers, R.C. Rhesus monkey rhadinovirus uses eph family receptors for entry into B cells and endothelial cells but not fibroblasts. PLoS Pathog. 2013, 9, e1003360. [Google Scholar] [CrossRef]
- Grosskopf, A.K.; Ensser, A.; Neipel, F.; Jungnickl, D.; Schlagowski, S.; Desrosiers, R.C.; Hahn, A.S. A conserved Eph family receptor-binding motif on the gH/gL complex of Kaposi’s sarcoma-associated herpesvirus and rhesus monkey rhadinovirus. PLoS Pathog. 2018, 14, e1006912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrigues, H.J.; Rubinchikova, Y.E.; Dipersio, C.M.; Rose, T.M. Integrin alphaVbeta3 Binds to the RGD motif of glycoprotein B of Kaposi’s sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J. Virol. 2008, 82, 1570–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrigues, H.J.; DeMaster, L.K.; Rubinchikova, Y.E.; Rose, T.M. Corrigendum to: “KSHV attachment and entry are dependent on alphaVbeta3 integrin localized to specific cell surface microdomains and do not correlate with the presence of heparan sulfate” [Virology 2014, 464–465, 118–133]. Virology 2018, 515, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Schaller, S.; Jardetzky, T.S.; Longnecker, R. Ephrin Receptor A4 is a New Kaposi’s Sarcoma-Associated Herpesvirus Virus Entry Receptor. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Rappocciolo, G.; Jenkins, F.J.; Hensler, H.R.; Piazza, P.; Jais, M.; Borowski, L.; Watkins, S.C.; Rinaldo, C.R., Jr. DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J. Immunol. 2006, 176, 1741–1749. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Sadagopan, S.; Bottero, V.; Paul, A.G.; Chandran, B. Characterization of entry and infection of monocytic THP-1 cells by Kaposi’s sarcoma associated herpesvirus (KSHV): Role of heparan sulfate, DC-SIGN, integrins and signaling. Virology 2010, 406, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Rappocciolo, G.; Hensler, H.R.; Jais, M.; Reinhart, T.A.; Pegu, A.; Jenkins, F.J.; Rinaldo, C.R. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J. Virol. 2008, 82, 4793–4806. [Google Scholar] [CrossRef] [Green Version]
- Grosskopf, A.K.; Schlagowski, S.; Hornich, B.F.; Fricke, T.; Desrosiers, R.C.; Hahn, A.S. EphA7 Functions as Receptor on BJAB Cells for Cell-to-Cell Transmission of the Kaposi’s Sarcoma-Associated Herpesvirus and for Cell-Free Infection by the Related Rhesus Monkey Rhadinovirus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 2009, 83, 4895–4911. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; ValiyaVeettil, M.; Sadagopan, S.; Paudel, N.; Chandran, B. c-Cbl-mediated selective virus-receptor translocations into lipid rafts regulate productive Kaposi’s sarcoma-associated herpesvirus infection in endothelial cells. J. Virol. 2011, 85, 12410–12430. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhou, F.; Greene, W.; Gao, S.J. Rhesus rhadinovirus infection of rhesus fibroblasts occurs through clathrin-mediated endocytosis. J. Virol. 2010, 84, 11709–11717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranatt, P.P.; Akula, S.M.; Zien, C.A.; Krishnan, H.H.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: Implications for infectivity. J. Virol. 2003, 77, 1524–1539. [Google Scholar] [CrossRef] [Green Version]
- Veettil, M.V.; Sharma-Walia, N.; Sadagopan, S.; Raghu, H.; Sivakumar, R.; Naranatt, P.P.; Chandran, B. RhoA-GTPase facilitates entry of Kaposi’s sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 2006, 80, 11432–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaleeba, J.A.; Berger, E.A. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: Cystine transporter xCT. Science 2006, 311, 1921–1924. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Chandran, B. KSHV Entry and Trafficking in Target Cells-Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses 2016, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Winter, J.; Lal, R.B.; Offermann, M.K.; Koyano, S. Characterization of entry mechanisms of human herpesvirus 8 by using an Rta-dependent reporter cell line. J. Virol. 2003, 77, 8147–8152. [Google Scholar] [CrossRef] [Green Version]
- Akula, S.M.; Naranatt, P.P.; Walia, N.S.; Wang, F.Z.; Fegley, B.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J. Virol. 2003, 77, 7978–7990. [Google Scholar] [CrossRef] [Green Version]
- Arii, J.; Kawaguchi, Y. The Role of HSV Glycoproteins in Mediating Cell Entry. Adv. Exp. Med. Biol. 2018, 1045, 3–21. [Google Scholar] [CrossRef]
- Sathiyamoorthy, K.; Chen, J.; Longnecker, R.; Jardetzky, T.S. The COMPLEXity in herpesvirus entry. Curr. Opin. Virol. 2017, 24, 97–104. [Google Scholar] [CrossRef]
- Weed, D.J.; Nicola, A.V. Herpes simplex virus Membrane Fusion. Adv. Anat. Embryol. Cell Biol. 2017, 223, 29–47. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes virus fusion and entry: A story with many characters. Viruses 2012, 4, 800–832. [Google Scholar] [CrossRef] [PubMed]
- Dollery, S.J.; Delboy, M.G.; Nicola, A.V. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 2010, 84, 3759–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weed, D.J.; Dollery, S.J.; Komala Sari, T.; Nicola, A.V. Acidic pH Mediates Changes in Antigenic and Oligomeric Conformation of Herpes Simplex Virus gB and Is a Determinant of Cell-Specific Entry. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannah, B.P.; Cairns, T.M.; Bender, F.C.; Whitbeck, J.C.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 2009, 83, 6825–6836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dollery, S.J.; Wright, C.C.; Johnson, D.C.; Nicola, A.V. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 2011, 85, 9964–9973. [Google Scholar] [CrossRef] [Green Version]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Stampfer, S.D.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 2010, 84, 12924–12933. [Google Scholar] [CrossRef] [Green Version]
- Pauk, J.; Huang, M.L.; Brodie, S.J.; Wald, A.; Koelle, D.M.; Schacker, T.; Celum, C.; Selke, S.; Corey, L. Mucosal shedding of human herpesvirus 8 in men. N. Engl. J. Med. 2000, 343, 1369–1377. [Google Scholar] [CrossRef]
- He, J.; Bhat, G.; Kankasa, C.; Chintu, C.; Mitchell, C.; Duan, W.; Wood, C. Seroprevalence of human herpesvirus 8 among Zambian women of childbearing age without Kaposi’s sarcoma (KS) and mother-child pairs with KS. J. Infect. Dis. 1998, 178, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Boshoff, C.; Schulz, T.F.; Kennedy, M.M.; Graham, A.K.; Fisher, C.; Thomas, A.; McGee, J.O.; Weiss, R.A.; O’Leary, J.J. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1995, 1, 1274–1278. [Google Scholar] [CrossRef]
- Ensoli, B.; Sgadari, C.; Barillari, G.; Sirianni, M.C.; Sturzl, M.; Monini, P. Biology of Kaposi’s sarcoma. Eur. J. Cancer 2001, 37, 1251–1269. [Google Scholar] [CrossRef]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Gen. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.W.; Trotter, M.W.; Lagos, D.; Bourboulia, D.; Henderson, S.; Makinen, T.; Elliman, S.; Flanagan, A.M.; Alitalo, K.; Boshoff, C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Gen. 2004, 36, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Jussila, L.; Valtola, R.; Partanen, T.A.; Salven, P.; Heikkila, P.; Matikainen, M.T.; Renkonen, R.; Kaipainen, A.; Detmar, M.; Tschachler, E.; et al. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 1998, 58, 1599–1604. [Google Scholar] [PubMed]
- Sakakibara, S.; Tosato, G. Viral interleukin-6: Role in Kaposi’s sarcoma-associated herpesvirus: Associated malignancies. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2011, 31, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Roth, W.K.; Brandstetter, H.; Sturzl, M. Cellular and molecular features of HIV-associated Kaposi’s sarcoma. AIDS 1992, 6, 895–913. [Google Scholar] [CrossRef]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Schafer, G.; Blumenthal, M.J.; Katz, A.A. Interaction of human tumor viruses with host cell surface receptors and cell entry. Viruses 2015, 7, 2592–2617. [Google Scholar] [CrossRef] [Green Version]
- Chandran, B.; Hutt-Fletcher, L. Gammaherpesviruses entry and early events during infection. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—An update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Kikuta, H.; Itakura, O.; Taneichi, K.; Kohno, M. Tropism of human herpesvirus 8 for peripheral blood lymphocytes in patients with Castleman’s disease. Br. J. Haematol. 1997, 99, 790–793. [Google Scholar] [CrossRef] [Green Version]
- Bechtel, J.T.; Liang, Y.; Hvidding, J.; Ganem, D. Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J. Virol. 2003, 77, 6474–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarman, E.; Mesri, E.A.; Gershengorn, M.C. Viral G protein-coupled receptor and Kaposi’s sarcoma: A model of paracrine neoplasia? J. Exp. Med. 2000, 191, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Traylen, C.M.; Akula, S.M. Cell membrane-bound Kaposi’s sarcoma-associated herpesvirus-encoded glycoprotein B promotes virus latency by regulating expression of cellular Egr-1. J. Biol. Chem. 2010, 285, 37491–37502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarousse, N.; Chandran, B.; Coscoy, L. Lack of heparan sulfate expression in B-cell lines: Implications for Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 infections. J. Virol. 2008, 82, 12591–12597. [Google Scholar] [CrossRef] [Green Version]
- Myoung, J.; Ganem, D. Infection of primary human tonsillar lymphoid cells by KSHV reveals frequent but abortive infection of T cells. Virology 2011, 413, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, S.E.; Seligmann, M. B-cell neoplasia in man. Lancet (London, England) 1974, 2, 1230–1233. [Google Scholar] [CrossRef]
- Campbell, D.M.; Rappocciolo, G.; Jenkins, F.J.; Rinaldo, C.R. Dendritic cells: Key players in human herpesvirus 8 infection and pathogenesis. Front. Microbiol. 2014, 5, 452. [Google Scholar] [CrossRef]
- Totonchy, J.; Osborn, J.M.; Chadburn, A.; Nabiee, R.; Argueta, L.; Mikita, G.; Cesarman, E. KSHV induces immunoglobulin rearrangements in mature B lymphocytes. PLoS Pathog. 2018, 14, e1006967. [Google Scholar] [CrossRef] [Green Version]
- Chadburn, A.; Hyjek, E.M.; Tam, W.; Liu, Y.; Rengifo, T.; Cesarman, E.; Knowles, D.M. Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV.; HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology 2008, 53, 513–524. [Google Scholar] [CrossRef]
- Jenner, R.G.; Maillard, K.; Cattini, N.; Weiss, R.A.; Boshoff, C.; Wooster, R.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA 2003, 100, 10399–10404. [Google Scholar] [CrossRef] [Green Version]
- Du, M.Q.; Liu, H.; Diss, T.C.; Ye, H.; Hamoudi, R.A.; Dupin, N.; Meignin, V.; Oksenhendler, E.; Boshoff, C.; Isaacson, P.G. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 2001, 97, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Dollery, S.J.; Santiago-Crespo, R.J.; Kardava, L.; Moir, S.; Berger, E.A. Efficient infection of a human B cell line with cell-free Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassman, L.M.; Ellison, T.J.; Kedes, D.H. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Invest. 2011, 121, 752–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myoung, J.; Ganem, D. Active lytic infection of human primary tonsillar B cells by KSHV and its noncytolytic control by activated CD4+ T cells. J. Clin. Invest. 2011, 121, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Chandran, B.; Bloomer, C.; Chan, S.R.; Zhu, L.; Goldstein, E.; Horvat, R. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by spliced transcripts. Virology 1998, 249, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.S.; Albrecht, J.C.; Birkmann, A.; Yaguboglu, S.; Lang, D.; Fleckenstein, B.; Neipel, F. The immunogenic glycoprotein gp35-37 of human herpesvirus 8 is encoded by open reading frame K8.1. J. Virol. 1998, 72, 6725–6731. [Google Scholar]
- Zhu, L.; Puri, V.; Chandran, B. Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology 1999, 262, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Renne, R.; Ganem, D.; Forghani, B. Human herpesvirus 8 glycoprotein K8.1: Expression, post-translational modification and localization analyzed by monoclonal antibody. J. Clin. Virol. 2000, 17, 127–136. [Google Scholar] [CrossRef]
- Engels, E.A.; Sinclair, M.D.; Biggar, R.J.; Whitby, D.; Ebbesen, P.; Goedert, J.J.; Gastwirth, J.L. Latent class analysis of human herpesvirus 8 assay performance and infection prevalence in sub-saharan Africa and Malta. Int. J. Cancer 2000, 88, 1003–1008. [Google Scholar] [CrossRef]
- Wang, F.Z.; Akula, S.M.; Pramod, N.P.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 2001, 75, 7517–7527. [Google Scholar] [CrossRef] [Green Version]
- Luna, R.E.; Zhou, F.; Baghian, A.; Chouljenko, V.; Forghani, B.; Gao, S.J.; Kousoulas, K.G. Kaposi’s sarcoma-associated herpesvirus glycoprotein K8.1 is dispensable for virus entry. J. Virol. 2004, 78, 6389–6398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, L.; Denekamp, L.; Knapp, A.; Auerbach, M.R.; Damania, B.; Desrosiers, R.C. The primary sequence of rhesus monkey rhadinovirus isolate 26–95: Sequence similarities to Kaposi’s sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 2000, 74, 3388–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWire, S.M.; McVoy, M.A.; Damania, B. Kinetics of expression of rhesus monkey rhadinovirus (RRV) and identification and characterization of a polycistronic transcript encoding the RRV Orf50/Rta, RRV R8, and R8.1 genes. J. Virol. 2002, 76, 9819–9831. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.M.; Kedes, D.H. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J. Virol. 2006, 80, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.P.; Janjua, N.J.; Pepper, S.D.; Bennion, G.; Mackett, M.; Allen, T.; Nash, A.A.; Arrand, J.R. Identification and characterization of murine gammaherpesvirus 68 gp150: A virion membrane glycoprotein. J. Virol. 1996, 70, 3528–3535. [Google Scholar] [PubMed]
- Machiels, B.; Lete, C.; de Fays, K.; Mast, J.; Dewals, B.; Stevenson, P.G.; Vanderplasschen, A.; Gillet, L. The bovine herpesvirus 4 Bo10 gene encodes a nonessential viral envelope protein that regulates viral tropism through both positive and negative effects. J. Virol. 2011, 85, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Means, R.E. Characterization of the Herpesvirus saimiri Orf51 protein. Virology 2004, 326, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Janz, A.; Oezel, M.; Kurzeder, C.; Mautner, J.; Pich, D.; Kost, M.; Hammerschmidt, W.; Delecluse, H.J. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 2000, 74, 10142–10152. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.D.; Neuhierl, B.; Baldwin, G.; Rickinson, A.B.; Delecluse, H.J. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7065–7070. [Google Scholar] [CrossRef] [Green Version]
- De Lima, B.D.; May, J.S.; Stevenson, P.G. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J. Virol. 2004, 78, 5103–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.P.; Silvia, O.J.; Atkin, I.M.; Hughes, D.J.; Ebrahimi, B.; Adler, H. In vivo function of a gammaherpesvirus virion glycoprotein: Influence on B-cell infection and mononucleosis. J. Virol. 2004, 78, 10449–10459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.; Adler, H.; Stevenson, P.G. Glycosaminoglycan interactions in murine gammaherpesvirus-68 infection. PLoS ONE 2007, 2, e347. [Google Scholar] [CrossRef] [PubMed]
- Machiels, B.; Stevenson, P.G.; Vanderplasschen, A.; Gillet, L. A gammaherpesvirus uses alternative splicing to regulate its tropism and its sensitivity to neutralization. PLoS Pathog. 2013, 9, e1003753. [Google Scholar] [CrossRef]
- Haan, K.M.; Lee, S.K.; Longnecker, R. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology 2001, 290, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Ogembo, J.G.; Kannan, L.; Ghiran, I.; Nicholson-Weller, A.; Finberg, R.W.; Tsokos, G.C.; Fingeroth, J.D. Human complement receptor type 1/CD35 is an Epstein-Barr Virus receptor. Cell Rep. 2013, 3, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.Z.; Akula, S.M.; Sharma-Walia, N.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J. Virol. 2003, 77, 3131–3147. [Google Scholar] [CrossRef] [Green Version]
- Glauser, D.L.; Milho, R.; Frederico, B.; May, J.S.; Kratz, A.S.; Gillet, L.; Stevenson, P.G. Glycoprotein B cleavage is important for murid herpesvirus 4 to infect myeloid cells. J. Virol. 2013, 87, 10828–10842. [Google Scholar] [CrossRef] [Green Version]
- Hensler, H.R.; Tomaszewski, M.J.; Rappocciolo, G.; Rinaldo, C.R.; Jenkins, F.J. Human herpesvirus 8 glycoprotein B binds the entry receptor DC-SIGN. Virus Res. 2014, 190, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Garrigues, H.J.; DeMaster, L.K.; Rubinchikova, Y.E.; Rose, T.M. KSHV attachment and entry are dependent on alphaVbeta3 integrin localized to specific cell surface microdomains and do not correlate with the presence of heparan sulfate. Virology 2014, 464–465, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.R.; Hussein, H.A.; Akula, S.M. Disintegrin-like domain of glycoprotein B regulates Kaposi’s sarcoma-associated herpesvirus infection of cells. J. Gen. Virol. 2014, 95, 1770–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Sanchez, E.; Altmeyer, R.; Amara, A.; Schwartz, O.; Fieschi, F.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Despres, P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003, 4, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.A.; de Witte, L.; Bolmstedt, A.; van Kooyk, Y.; Geijtenbeek, T.B. Dendritic cells mediate herpes simplex virus infection and transmission through the C-type lectin DC-SIGN. J. Gen. Virol. 2008, 89, 2398–2409. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef] [Green Version]
- Sathiyamoorthy, K.; Jiang, J.; Hu, Y.X.; Rowe, C.L.; Mohl, B.S.; Chen, J.; Jiang, W.; Mellins, E.D.; Longnecker, R.; Zhou, Z.H.; et al. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog. 2014, 10, e1004309. [Google Scholar] [CrossRef] [Green Version]
- Hannah, B.P.; Heldwein, E.E.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 2007, 81, 4858–4865. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.; Birkmann, A.; Wies, E.; Dorer, D.; Mahr, K.; Sturzl, M.; Titgemeyer, F.; Neipel, F. Kaposi’s sarcoma-associated herpesvirus gH/gL: Glycoprotein export and interaction with cellular receptors. J. Virol. 2009, 83, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Pitulescu, M.E.; Adams, R.H. Eph/ephrin molecules—A hub for signaling and endocytosis. Genes Dev. 2010, 24, 2480–2492. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Chakraborty, S.; Bandyopadhyay, C.; Valiya Veettil, M.; Ansari, M.A.; Singh, V.V.; Chandran, B. EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS Pathog. 2013, 9, e1003510. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Veettil, M.V.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. USA 2012, 109, E1163–E1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omerovic, J.; Longnecker, R. Functional homology of gHs and gLs from EBV-related gamma-herpesviruses for EBV-induced membrane fusion. Virology 2007, 365, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, H.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. USA 2010, 107, 22641–22646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, J.; Ortego, M.S.; Weidle, C.; Stuart, A.B.; Gray, M.D.; McElrath, M.J.; Pancera, M.; Veesler, D.; McGuire, A.T. An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 2018, 48, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Spiller, O.B.; Robinson, M.; O’Donnell, E.; Milligan, S.; Morgan, B.P.; Davison, A.J.; Blackbourn, D.J. Complement regulation by Kaposi’s sarcoma-associated herpesvirus ORF4 protein. J. Virol. 2003, 77, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Myoung, J.; Ganem, D. Infection of lymphoblastoid cell lines by Kaposi’s sarcoma-associated herpesvirus: Critical role of cell-associated virus. J. Virol. 2011, 85, 9767–9777. [Google Scholar] [CrossRef] [Green Version]
- Pertel, P.E. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 2002, 76, 4390–4400. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, N.; Li, W.; Zhu, F.; Wang, Y.; Yuan, Y. Mono-ubiquitylated ORF45 Mediates Association of KSHV Particles with Internal Lipid Rafts for Viral Assembly and Egress. PLoS Pathog. 2015, 11, e1005332. [Google Scholar] [CrossRef]
- Albecka, A.; Laine, R.F.; Janssen, A.F.; Kaminski, C.F.; Crump, C.M. HSV-1 Glycoproteins Are Delivered to Virus Assembly Sites Through Dynamin-Dependent Endocytosis. Traffic (Copenhagen, Denmark) 2016, 17, 21–39. [Google Scholar] [CrossRef] [Green Version]
- Muggeridge, M.I. Glycoprotein B of herpes simplex virus 2 has more than one intracellular conformation and is altered by low pH. J. Virol. 2012, 86, 6444–6456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitia Ortiz de Zarate, I.; Cantero-Aguilar, L.; Longo, M.; Berlioz-Torrent, C.; Rozenberg, F. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J. Virol. 2007, 81, 13889–13903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, W.; Gao, S.J. Actin dynamics regulate multiple endosomal steps during Kaposi’s sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog. 2009, 5, e1000512. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Virion Glycoprotein | |||
|---|---|---|---|
| Cell Type | gB | gHgL | K8.1A |
| Endothelial | α3β1 [38,39], αVβ3 [39], αVβ5 [39] | EphA2 [40,41,42] | |
| Epithelial | α3β1 [39], αVβ3 [39,43,44], αVβ5 [39] * | EphA2 [40,41,42] EphA4 [45] | |
| Fibroblasts | α3β1 [38,39], αVβ3 [39], αVβ5 [39] | ||
| Monocytes | DC-SIGN [46,47], α3β1 [47], αVβ3 [47], αVβ5 [47], αVβ1 [47] | ||
| Macrophages | DC-SIGN [46] | ||
| Dendritic cells | DC-SIGN [46] | ||
| B cells | DC-SIGN (activated B cells) [48] | EphA7 in cell–cell spread [49] * | K8.1A needed [34] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dollery, S.J. Towards Understanding KSHV Fusion and Entry. Viruses 2019, 11, 1073. https://doi.org/10.3390/v11111073
Dollery SJ. Towards Understanding KSHV Fusion and Entry. Viruses. 2019; 11(11):1073. https://doi.org/10.3390/v11111073
Chicago/Turabian StyleDollery, Stephen J. 2019. "Towards Understanding KSHV Fusion and Entry" Viruses 11, no. 11: 1073. https://doi.org/10.3390/v11111073
APA StyleDollery, S. J. (2019). Towards Understanding KSHV Fusion and Entry. Viruses, 11(11), 1073. https://doi.org/10.3390/v11111073