The Evolutionary Arms Race between Virus and NK Cells: Diversity Enables Population-Level Virus Control



Abstract
1. Introduction
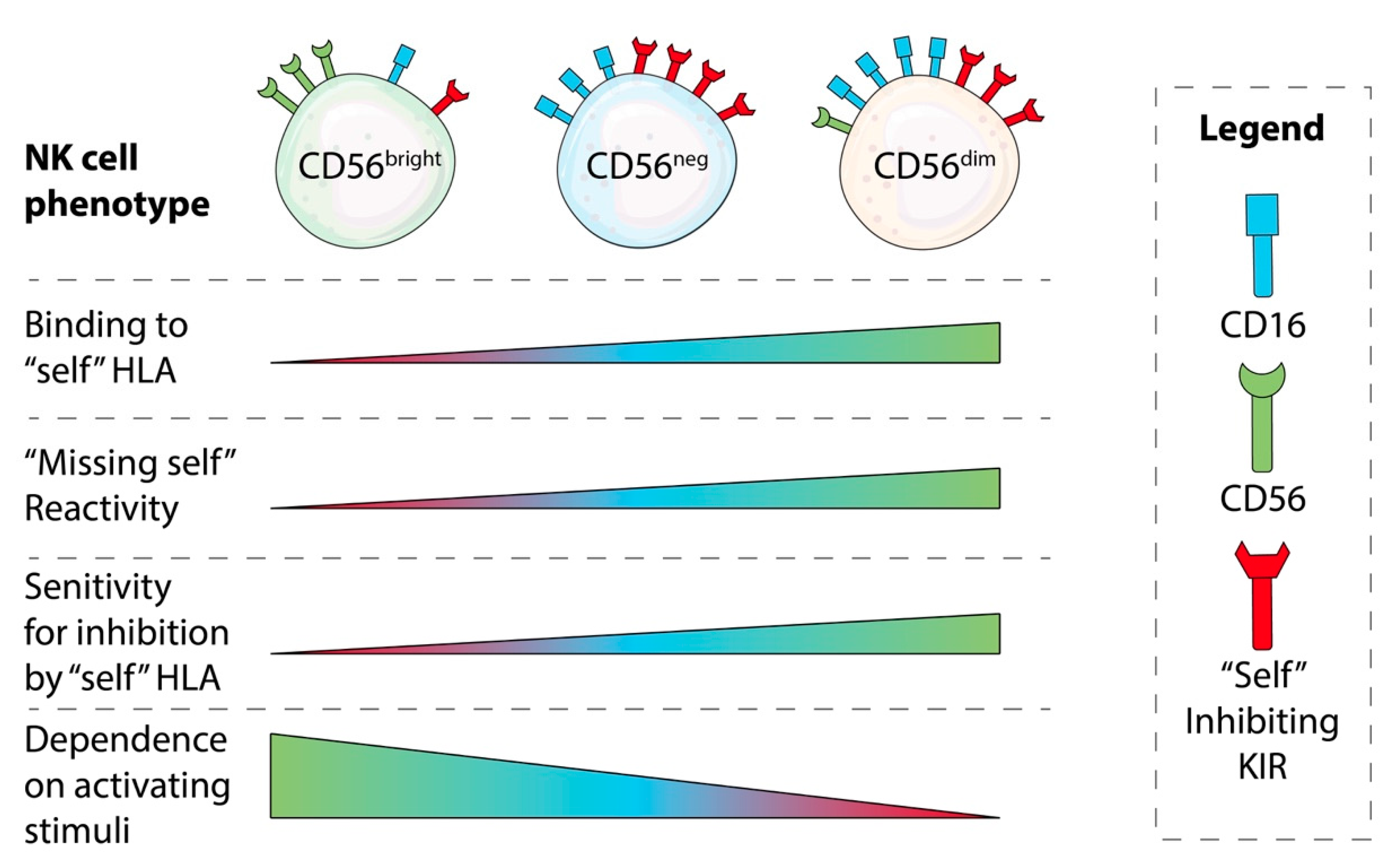
NK Cell Education: Impacts of Diverse Immunogenetics
2. Taking a Calculated Risk: Genetic Variation between Hosts Creates an Opportunity for a Variety of Infection Outcomes
2.1. HIV Induces HLA-Bw4 Downregulation, Creating a Target for Educated KIR3DL1+ NK Cells
2.2. Downregulation of HLA-C by HIV Creates a Target for NK Cells Educated by KIR2DL1/2/3
2.3. Immunogenetic Diversity Predicts Susceptibility, Resistance and Disease Processes in Hepatitis C Virus Infection
2.4. The KIR Strike Back: Activating KIR Are Associated with Virus Control
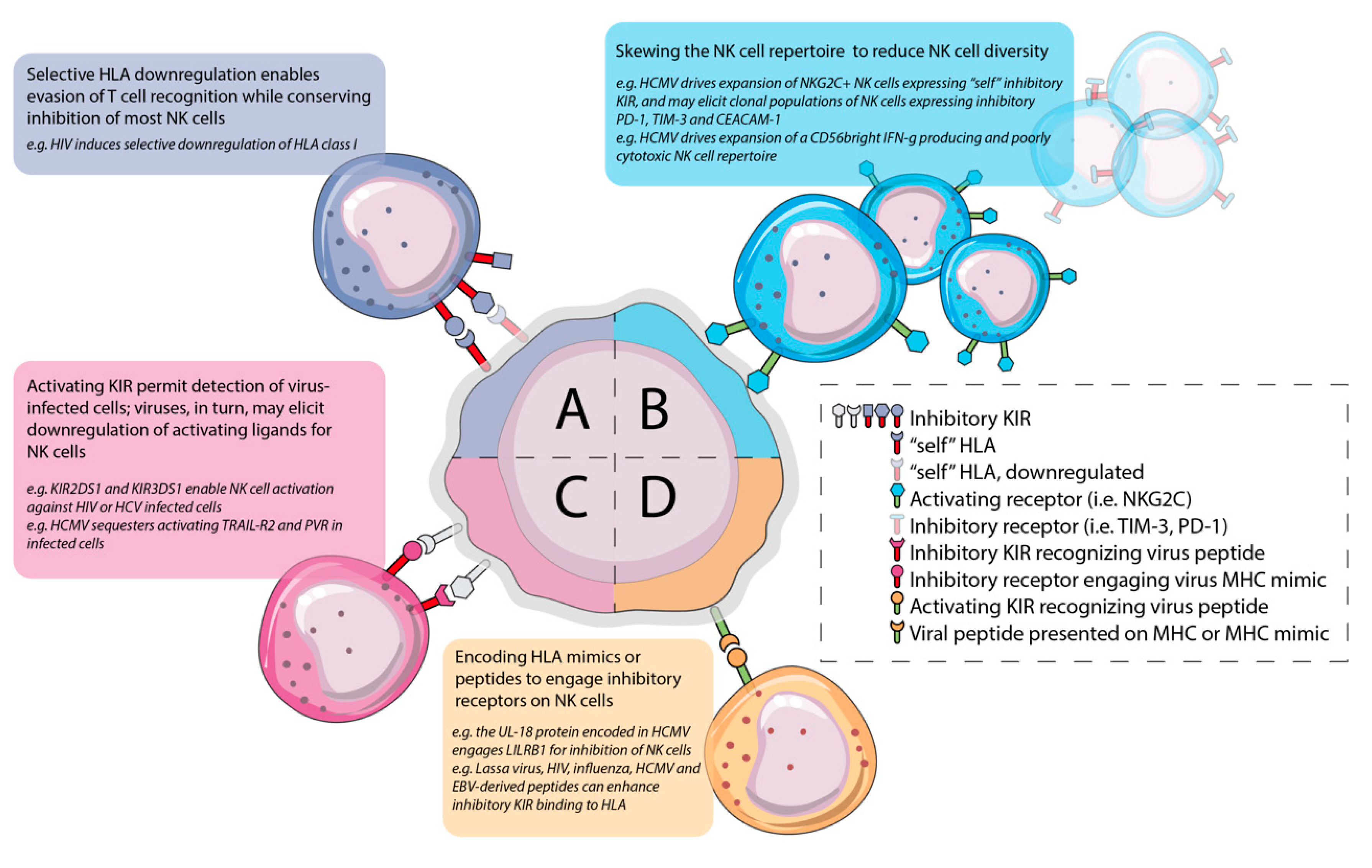
3. Distract and Redirect: Infection Leaves Stable Imprints on the NK Cell Repertoire that Impact Viral Control and Enable Chronic Infection
3.1. HCMV Infection Drives Expansion of Adaptive/Memory Self-KIR+ NKG2C+ NK Populations and May Impact the Outcome of Co-Infection
3.2. Chronic HCMV Infection is Associated with Dysfunction and Exhaustion in NK Cells
4. Resetting the Balance to Favor Inhibition over Activation
4.1. Chronic Infection Induces Inhibitory Receptor Expression and NK Cell Exhaustion or Diminishes Activating Receptor Expression
4.2. Virally-Encoded Peptides Alter KIR-HLA Interactions to Favour Inhibition
5. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Voss, M.; Bryceson, Y.T. Natural killer cell biology illuminated by primary immunodeficiency syndromes in humans. Clin. Immunol. 2017, 177, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Hsu, K.C. Natural Killer Cell Education and the Response to Infection and Cancer Therapy: Stay Tuned. Trends Immunol. 2018, 39, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Karre, K.; Hoglund, P. NK cell education: Not an on-off switch but a tunable rheostat. Trends Immunol. 2009, 30, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef]
- Pahl, J.H.W.; Cerwenka, A.; Ni, J. Memory-Like NK Cells: Remembering a Previous Activation by Cytokines and NK Cell Receptors. Front Immunol. 2018, 9, 2796. [Google Scholar] [CrossRef]
- Karre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Abi-Rached, L.; Parham, P. Natural selection drives recurrent formation of activating killer cell immunoglobulin-like receptor and Ly49 from inhibitory homologues. J. Exp. Med. 2005, 201, 1319–1332. [Google Scholar] [CrossRef]
- Kelley, J.; Walter, L.; Trowsdale, J. Comparative genomics of natural killer cell receptor gene clusters. PLoS Genet. 2005, 1, 129–139. [Google Scholar] [CrossRef]
- Carrillo-Bustamante, P.; Kesmir, C.; de Boer, R.J. The evolution of natural killer cell receptors. Immunogenetics 2016, 68, 3–18. [Google Scholar] [CrossRef]
- Raulet, D.H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 2006, 18, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sunwoo, J.B.; Yang, L.; Choi, T.; Song, Y.J.; French, A.R.; Vlahiotis, A.; Piccirillo, J.F.; Cella, M.; Colonna, M.; et al. HLA alleles determine differences in human natural killer cell responsiveness and potency. Proc. Natl. Acad. Sci. USA 2008, 105, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Johnson, C.; Jayaraman, J.; Simecek, N.; Noble, J.; Moffatt, M.F.; Cookson, W.O.; Trowsdale, J.; Traherne, J.A. Copy number variation leads to considerable diversity for B but not A haplotypes of the human KIR genes encoding NK cell receptors. Genome Res. 2012, 22, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Vierra-Green, C.; Roe, D.; Jayaraman, J.; Trowsdale, J.; Traherne, J.; Kuang, R.; Spellman, S.; Maiers, M. Estimating KIR Haplotype Frequencies on a Cohort of 10,000 Individuals: A Comprehensive Study on Population Variations, Typing Resolutions, and Reference Haplotypes. PLoS ONE 2016, 11, e0163973. [Google Scholar] [CrossRef]
- Hsu, K.C.; Liu, X.R.; Selvakumar, A.; Mickelson, E.; O’Reilly, R.J.; Dupont, B. Killer Ig-Like Receptor Haplotype Analysis by Gene Content: Evidence for Genomic Diversity with a Minimum of Six Basic Framework Haplotypes, Each with Multiple Subsets. J. Immunol. 2002, 169, 5118–5129. [Google Scholar] [CrossRef]
- Wroblewski, E.E.; Parham, P.; Guethlein, L.A. Two to Tango: Co-Evolution of Hominid Natural Killer Cell Receptors and MHC. Front. Immunol. 2019, 10, 177. [Google Scholar] [CrossRef]
- Thomas, R.; Yamada, E.; Alter, G.; Martin, M.P.; Bashirova, A.A.; Norman, P.J.; Altfeld, M.; Parham, P.; Anderson, S.K.; McVicar, D.W.; et al. Novel KIR3DL1 Alleles and Their Expression Levels on NK Cells: Convergent Evolution of KIR3DL1 Phenotype Variation? J. Immunol. 2008, 180, 6743–6750. [Google Scholar] [CrossRef]
- Parham, P.; Norman, P.J.; Abi-Rached, L.; Guethlein, L.A. Variable NK cell receptors exemplified by human KIR3DL1/S1. J. Immunol. 2011, 187, 11–19. [Google Scholar] [CrossRef]
- Boudreau, J.E.; Mulrooney, T.J.; Le Luduec, J.B.; Barker, E.; Hsu, K.C. KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV. J. Immunol. 2016, 196, 3398–3410. [Google Scholar] [CrossRef]
- Brodin, P.; Hoglund, P. Beyond licensing and disarming: A quantitative view on NK-cell education. Eur. J. Immunol. 2008, 38, 2934–2937. [Google Scholar] [CrossRef]
- Brodin, P.; Lakshmikanth, T.; Johansson, S.; Karre, K.; Hoglund, P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood 2009, 113, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Peng, H.; Sun, R.; Wei, H.; Ljunggren, H.G.; Yokoyama, W.M.; Tian, Z. Contribution of inhibitory receptor TIGIT to NK cell education. J. Autoimmun. 2017, 81, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yawata, M.; Yawata, N.; Draghi, M.; Little, A.M.; Partheniou, F.; Parham, P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J. Exp. Med. 2006, 203, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Hilton, H.G.; Parham, P. Missing or altered self: Human NK cell receptors that recognize HLA-C. Immunogenetics 2017, 69, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Mwimanzi, P.; Markle, T.J.; Ogata, Y.; Martin, E.; Tokunaga, M.; Mahiti, M.; Kuang, X.T.; Walker, B.D.; Brockman, M.A.; Brumme, Z.L.; et al. Dynamic range of Nef functions in chronic HIV-1 infection. Virology 2013, 439, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Bonaparte, M.I.; Barker, E. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood 2004, 104, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Mwimanzi, F.; Toyoda, M.; Mahiti, M.; Mann, J.K.; Martin, J.N.; Bangsberg, D.; Brockman, M.A.; Goulder, P.; Kirchhoff, F.; Brumme, Z.L.; et al. Resistance of Major Histocompatibility Complex Class B (MHC-B) to Nef-Mediated Downregulation Relative to that of MHC-A Is Conserved among Primate Lentiviruses and Influences Antiviral T Cell Responses in HIV-1-Infected Individuals. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Jia, X.; Singh, R.; Homann, S.; Yang, H.; Guatelli, J.; Xiong, Y. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 2012, 19, 701–706. [Google Scholar] [CrossRef]
- Davis, Z.B.; Sowrirajan, B.; Cogswell, A.; Ward, J.P.; Planelles, V.; Barker, E. CD155 on HIV-Infected Cells Is Not Modulated by HIV-1 Vpu and Nef but Synergizes with NKG2D Ligands to Trigger NK Cell Lysis of Autologous Primary HIV-Infected Cells. AIDS Res. Hum. Retrovir. 2017, 33, 93–100. [Google Scholar] [CrossRef]
- Saunders, P.M.; Pymm, P.; Pietra, G.; Hughes, V.A.; Hitchen, C.; O’Connor, G.M.; Loiacono, F.; Widjaja, J.; Price, D.A.; Falco, M.; et al. Killer cell immunoglobulin-like receptor 3DL1 polymorphism defines distinct hierarchies of HLA class I recognition. J. Exp. Med. 2016, 213, 791–807. [Google Scholar] [CrossRef]
- Boudreau, J.E.; Giglio, F.; Gooley, T.A.; Stevenson, P.A.; Le Luduec, J.B.; Shaffer, B.C.; Rajalingam, R.; Hou, L.; Hurley, C.K.; Noreen, H.; et al. KIR3DL1/ HL A-B Subtypes Govern Acute Myelogenous Leukemia Relapse After Hematopoietic Cell Transplantation. J. Clin. Oncol. 2017, 35, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Taner, S.B.; Pando, M.J.; Roberts, A.; Schellekens, J.; Marsh, S.G.; Malmberg, K.J.; Parham, P.; Brodsky, F.M. Interactions of NK cell receptor KIR3DL1*004 with chaperones and conformation-specific antibody reveal a functional folded state as well as predominant intracellular retention. J. Immunol. 2011, 186, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Gao, X.; Lee, J.H.; Nelson, G.W.; Detels, R.; Goedert, J.J.; Buchbinder, S.; Hoots, K.; Vlahov, D.; Trowsdale, J.; et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat. Genet. 2002, 31, 429–434. [Google Scholar] [CrossRef]
- Korner, C.; Simoneau, C.R.; Schommers, P.; Granoff, M.; Ziegler, M.; Holzemer, A.; Lunemann, S.; Chukwukelu, J.; Corleis, B.; Naranbhai, V.; et al. HIV-1-Mediated Downmodulation of HLA-C Impacts Target Cell Recognition and Antiviral Activity of NK Cells. Cell Host Microbe 2017, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Del Prete, G.Q.; Chatterjee, P.; Lara, A.; Brumme, Z.L.; Brockman, M.A.; Neil, S.; Pickering, S.; Schneider, D.K.; Piechocka-Trocha, A.; et al. HIV-1 Vpu Mediates HLA-C Downregulation. Cell Host Microbe 2016, 19, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.K. Molecular evolution of elements controlling HLA-C expression: Adaptation to a role as a killer-cell immunoglobulin-like receptor ligand regulating natural killer cell function. HLA 2018, 92, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hilton, H.G.; Guethlein, L.A.; Goyos, A.; Nemat-Gorgani, N.; Bushnell, D.A.; Norman, P.J.; Parham, P. Polymorphic HLA-C Receptors Balance the Functional Characteristics of KIR Haplotypes. J. Immunol. 2015, 195, 3160–3170. [Google Scholar] [CrossRef]
- Moesta, A.K.; Parham, P. Diverse functionality among human NK cell receptors for the C1 epitope of HLA-C: KIR2DS2, KIR2DL2, and KIR2DL3. Front. Immunol. 2012, 3, 336. [Google Scholar] [CrossRef]
- Frazier, W.R.; Steiner, N.; Hou, L.; Dakshanamurthy, S.; Hurley, C.K. Allelic variation in KIR2DL3 generates a KIR2DL2-like receptor with increased binding to its HLA-C ligand. J. Immunol. 2013, 190, 6198–6208. [Google Scholar] [CrossRef]
- David, G.; Djaoud, Z.; Willem, C.; Legrand, N.; Rettman, P.; Gagne, K.; Cesbron, A.; Retiere, C. Large spectrum of HLA-C recognition by killer Ig-like receptor (KIR)2DL2 and KIR2DL3 and restricted C1 SPECIFICITY of KIR2DS2: Dominant impact of KIR2DL2/KIR2DS2 on KIR2D NK cell repertoire formation. J. Immunol. 2013, 191, 4778–4788. [Google Scholar] [CrossRef]
- Apps, R.; Qi, Y.; Carlson, J.M.; Chen, H.; Gao, X.; Thomas, R.; Yuki, Y.; Del Prete, G.Q.; Goulder, P.; Brumme, Z.L.; et al. Influence of HLA-C expression level on HIV control. Science 2013, 340, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Le Luduec, J.B.; Boudreau, J.E.; Freiberg, J.C.; Hsu, K.C. Novel Approach to Cell Surface Discrimination Between KIR2DL1 Subtypes and KIR2DS1 Identifies Hierarchies in NK Repertoire, Education, and Tolerance. Front. Immunol. 2019, 10, 734. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, S.I.; Thio, C.L.; Martin, M.P.; Brooks, C.R.; Gao, X.; Astemborski, J.; Cheng, J.; Goedert, J.J.; Vlahov, D.; Hilgartner, M.; et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 2004, 305, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Warshow, U.; Hegazy, D.; Brackenbury, L.; Guha, I.N.; Fowell, A.; Little, A.M.; Alexander, G.J.; Rosenberg, W.M.; Cramp, M.E.; et al. Consistent beneficial effects of killer cell immunoglobulin-like receptor 2DL3 and group 1 human leukocyte antigen-C following exposure to hepatitis C virus. Hepatology 2010, 51, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Gauthiez, E.; Habfast-Robertson, I.; Rueger, S.; Kutalik, Z.; Aubert, V.; Berg, T.; Cerny, A.; Gorgievski, M.; George, J.; Heim, M.H.; et al. A systematic review and meta-analysis of HCV clearance. Liver Int. 2017, 37, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Gaudieri, S.; Armstrong, N.J.; O’Connor, K.S.; Berg, T.; Weltman, M.; Abate, M.L.; Spengler, U.; Bassendine, M.; Dore, G.J.; et al. IL28B, HLA-C, and KIR variants additively predict response to therapy in chronic hepatitis C virus infection in a European Cohort: A cross-sectional study. PLoS Med. 2011, 8, e1001092. [Google Scholar] [CrossRef]
- Vidal-Castineira, J.R.; Lopez-Vazquez, A.; Diaz-Pena, R.; Alonso-Arias, R.; Martinez-Borra, J.; Perez, R.; Fernandez-Suarez, J.; Melon, S.; Prieto, J.; Rodrigo, L.; et al. Effect of killer immunoglobulin-like receptors in the response to combined treatment in patients with chronic hepatitis C virus infection. J. Virol. 2010, 84, 475–481. [Google Scholar] [CrossRef]
- Thoens, C.; Heinold, A.; Lindemann, M.; Horn, P.A.; Chang, D.I.; Scherbaum, N.; Timm, J.; Heinemann, F.M. A Single-Nucleotide Polymorphism Upstream of the HLA-C Locus Is Associated with an Anti-Hepatitis C Virus-Seronegative State in a High-Risk Exposed Cohort. J. Infect. Dis 2018, 218, 2016–2019. [Google Scholar] [CrossRef]
- Thons, C.; Senff, T.; Hydes, T.J.; Manser, A.R.; Heinemann, F.M.; Heinold, A.; Heilmann, M.; Kim, A.Y.; Uhrberg, M.; Scherbaum, N.; et al. HLA-Bw4 80(T) and multiple HLA-Bw4 copies combined with KIR3DL1 associate with spontaneous clearance of HCV infection in people who inject drugs. J. Hepatol. 2017, 67, 462–470. [Google Scholar] [CrossRef]
- Fauriat, C.; Ivarsson, M.A.; Ljunggren, H.G.; Malmberg, K.J.; Michaelsson, J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood 2010, 115, 1166–1174. [Google Scholar] [CrossRef]
- Pittari, G.; Liu, X.R.; Selvakumar, A.; Zhao, Z.; Merino, E.; Huse, M.; Chewning, J.H.; Hsu, K.C.; Dupont, B. NK cell tolerance of self-specific activating receptor KIR2DS1 in individuals with cognate HLA-C2 ligand. J. Immunol. 2013, 190, 4650–4660. [Google Scholar] [CrossRef] [PubMed]
- Burian, A.; Wang, K.L.; Finton, K.A.; Lee, N.; Ishitani, A.; Strong, R.K.; Geraghty, D.E. HLA-F and MHC-I Open Conformers Bind Natural Killer Cell Ig-Like Receptor KIR3DS1. PLoS ONE 2016, 11, e0163297. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, S.; Schobel, A.; Kah, J.; Fittje, P.; Holzemer, A.; Langeneckert, A.E.; Hess, L.U.; Poch, T.; Martrus, G.; Garcia-Beltran, W.F.; et al. Interactions Between KIR3DS1 and HLA-F Activate Natural Killer Cells to Control HCV Replication in Cell Culture. Gastroenterology 2018, 155, 1366–1371 e1363. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Holzemer, A.; Martrus, G.; Chung, A.W.; Pacheco, Y.; Simoneau, C.R.; Rucevic, M.; Lamothe-Molina, P.A.; Pertel, T.; Kim, T.E.; et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol. 2016, 17, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; Caggiari, L.; de Zorzi, M.; Repetto, O.; Zignego, A.L.; Izzo, F.; Tornesello, M.L.; Buonaguro, F.M.; Mangia, A.; Sansonno, D.; et al. Genetic diversity of the KIR/HLA system and susceptibility to hepatitis C virus-related diseases. PLoS ONE 2015, 10, e0117420. [Google Scholar] [CrossRef]
- Lopez-Vazquez, A.; Rodrigo, L.; Martinez-Borra, J.; Perez, R.; Rodriguez, M.; Fdez-Morera, J.L.; Fuentes, D.; Rodriguez-Rodero, S.; Gonzaez, S.; Lopez-Larrea, C. Protective effect of the HLA-Bw4I80 epitope and the killer cell immunoglobulin-like receptor 3DS1 gene against the development of hepatocellular carcinoma in patients with hepatitis C virus infection. J. Infect. Dis. 2005, 192, 162–165. [Google Scholar] [CrossRef]
- Malnati, M.S.; Ugolotti, E.; Monti, M.C.; Battista, D.; Vanni, I.; Bordo, D.; Sironi, F.; Larghero, P.; Marco, E.D.; Biswas, P.; et al. Activating Killer Immunoglobulin Receptors and HLA-C: A successful combination providing HIV-1 control. Sci. Rep. 2017, 7, 42470. [Google Scholar] [CrossRef]
- Dulberger, C.L.; McMurtrey, C.P.; Holzemer, A.; Neu, K.E.; Liu, V.; Steinbach, A.M.; Garcia-Beltran, W.F.; Sulak, M.; Jabri, B.; Lynch, V.J.; et al. Human Leukocyte Antigen F Presents Peptides and Regulates Immunity through Interactions with NK Cell Receptors. Immunity 2017, 46, 1018–1029. [Google Scholar] [CrossRef]
- Kiani, Z.; Bruneau, J.; Geraghty, D.E.; Bernard, N.F. HLA-F on Autologous HIV-Infected Cells Activates Primary NK Cells Expressing the Activating Killer Immunoglobulin-Like Receptor KIR3DS1. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Mele, D.; Pasi, A.; Cacciatore, R.; Mantovani, S.; Oliviero, B.; Mondelli, M.U.; Varchetta, S. Decreased interferon-gamma production by NK cells from KIR haplotype B carriers in hepatitis C virus infection. Liver Int. 2019, 39, 1237–1245. [Google Scholar] [CrossRef]
- Vendrame, E.; Fukuyama, J.; Strauss-Albee, D.M.; Holmes, S.; Blish, C.A. Mass Cytometry Analytical Approaches Reveal Cytokine-Induced Changes in Natural Killer Cells. Cytom. B Clin. Cytom. 2017, 92, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Blish, C.A. Diversification of human NK cells: Lessons from deep profiling. J. Leukoc. Biol. 2018, 103, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Vilches, C.; Angulo, A.; Lopez-Botet, M. Adaptive reconfiguration of the human NK-cell compartment in response to cytomegalovirus: A different perspective of the host-pathogen interaction. Eur. J. Immunol. 2013, 43, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef]
- Adams, E.J.; Juo, Z.S.; Venook, R.T.; Boulanger, M.J.; Arase, H.; Lanier, L.L.; Garcia, K.C. Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10128–10133. [Google Scholar] [CrossRef]
- Fodil-Cornu, N.; Lee, S.H.; Belanger, S.; Makrigiannis, A.P.; Biron, C.A.; Buller, R.M.; Vidal, S.M. Ly49h-deficient C57BL/6 mice: A new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol. 2008, 181, 6394–6405. [Google Scholar] [CrossRef]
- Mitrovic, M.; Arapovic, J.; Jordan, S.; Fodil-Cornu, N.; Ebert, S.; Vidal, S.M.; Krmpotic, A.; Reddehase, M.J.; Jonjic, S. The NK cell response to mouse cytomegalovirus infection affects the level and kinetics of the early CD8(+) T-cell response. J. Virol. 2012, 86, 2165–2175. [Google Scholar] [CrossRef]
- Pyzik, M.; Kielczewska, A.; Vidal, S.M. NK cell receptors and their MHC class I ligands in host response to cytomegalovirus: Insights from the mouse genome. Semin. Immunol. 2008, 20, 331–342. [Google Scholar] [CrossRef]
- Beziat, V.; Dalgard, O.; Asselah, T.; Halfon, P.; Bedossa, P.; Boudifa, A.; Hervier, B.; Theodorou, I.; Martinot, M.; Debre, P.; et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur. J. Immunol. 2012, 42, 447–457. [Google Scholar] [CrossRef]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef]
- Cao, K.; Marin, D.; Sekine, T.; Rondon, G.; Zhao, W.; Smith, N.T.; Daher, M.; Wang, Q.; Li, L.; Saliba, R.M.; et al. Donor NKG2C Copy Number: An Independent Predictor for CMV Reactivation After Double Cord Blood Transplantation. Front. Immunol. 2018, 9, 2444. [Google Scholar] [CrossRef]
- Liu, L.L.; Landskron, J.; Ask, E.H.; Enqvist, M.; Sohlberg, E.; Traherne, J.A.; Hammer, Q.; Goodridge, J.P.; Larsson, S.; Jayaraman, J.; et al. Critical Role of CD2 Co-stimulation in Adaptive Natural Killer Cell Responses Revealed in NKG2C-Deficient Humans. Cell Rep. 2016, 15, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Comeau, E.M.; Holder, K.A.; Fudge, N.J.; Grant, M.D. Cytomegalovirus-Driven Adaption of Natural Killer Cells in NKG2C(null) Human Immunodeficiency Virus-Infected Individuals. Viruses 2019, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Makwana, N.B.; Foley, B.; Lee, S.; Fernandez, S.; Irish, A.B.; Price, P. Asymptomatic CMV infections in long-term renal transplant recipients are associated with the loss of FcRgamma from LIR-1(+) NK cells. Eur. J. Immunol. 2016, 46, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; et al. CD56dimCD57+NKG2C+NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Taras, E.; Chiuppesi, F.; Wagner, J.E.; Blazar, B.R.; Brunstein, C.; Luo, X.; Diamond, D.J.; Cooley, S.; Weisdorf, D.J.; et al. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Peppa, D.; Pedroza-Pacheco, I.; Pellegrino, P.; Williams, I.; Maini, M.K.; Borrow, P. Adaptive Reconfiguration of Natural Killer Cells in HIV-1 Infection. Front. Immunol. 2018, 9, 474. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015, 42, 431–442. [Google Scholar] [CrossRef]
- Hwang, I.; Zhang, T.; Scott, J.M.; Kim, A.R.; Lee, T.; Kakarla, T.; Kim, A.; Sunwoo, J.B.; Kim, S. Identification of human NK cells that are deficient for signaling adaptor FcRgamma and specialized for antibody-dependent immune functions. Int. Immunol. 2012, 24, 793–802. [Google Scholar] [CrossRef]
- Zhang, T.; Scott, J.M.; Hwang, I.; Kim, S. Cutting edge: Antibody-dependent memory-like NK cells distinguished by FcRgamma deficiency. J. Immunol. 2013, 190, 1402–1406. [Google Scholar] [CrossRef]
- Oh, J.S.; Ali, A.K.; Kim, S.; Corsi, D.J.; Cooper, C.L.; Lee, S.H. NK cells lacking FcepsilonRIgamma are associated with reduced liver damage in chronic hepatitis C virus infection. Eur. J. Immunol. 2016, 46, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Kared, H.; Martelli, S.; Tan, S.W.; Simoni, Y.; Chong, M.L.; Yap, S.H.; Newell, E.W.; Pender, S.L.F.; Kamarulzaman, A.; Rajasuriar, R.; et al. Adaptive NKG2C(+)CD57(+) Natural Killer Cell and Tim-3 Expression During Viral Infections. Front. Immunol. 2018, 9, 686. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346 e333. [Google Scholar] [CrossRef] [PubMed]
- Peppa, D. Natural Killer Cells in Human Immunodeficiency Virus-1 Infection: Spotlight on the Impact of Human Cytomegalovirus. Front. Immunol. 2017, 8, 1322. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.; Ahmad, F.; Wedemeyer, H.; Cornberg, M.; Schulze Zur Wiesch, J.; van Lunzen, J.; Sarin, S.K.; Schmidt, R.E.; Meyer-Olson, D. Increased CD56(bright) NK cells in HIV-HCV co-infection and HCV mono-infection are associated with distinctive alterations of their phenotype. Virol. J. 2016, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Tanimine, N.; Tanaka, Y.; Abe, T.; Piao, J.; Chayama, K.; Ohdan, H. Functional Behavior of NKp46-Positive Intrahepatic Natural Killer Cells Against Hepatitis C Virus Reinfection After Liver Transplantation. Transplantation 2016, 100, 355–364. [Google Scholar] [CrossRef]
- Ortega-Prieto, A.M.; Dorner, M. Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection. Vaccines 2017, 5, 24. [Google Scholar] [CrossRef]
- Mina, M.M.; Cameron, B.; Luciani, F.; Vollmer-Conna, U.; Lloyd, A.R. Natural killer cells in highly exposed hepatitis C-seronegative injecting drug users. J. Viral Hepat. 2016, 23, 464–472. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Falco, M.; Podesta, M.; Locatelli, F.; Moretta, L.; Frassoni, F.; Moretta, A. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: A role for human cytomegalovirus? Blood 2012, 119, 399–410. [Google Scholar] [CrossRef]
- Gonzalez, V.D.; Falconer, K.; Bjorkstrom, N.K.; Blom, K.G.; Weiland, O.; Ljunggren, H.G.; Alaeus, A.; Sandberg, J.K. Expansion of functionally skewed CD56-negative NK cells in chronic hepatitis C virus infection: Correlation with outcome of pegylated IFN-alpha and ribavirin treatment. J. Immunol. 2009, 183, 6612–6618. [Google Scholar] [CrossRef]
- Strunz, B.; Hengst, J.; Deterding, K.; Manns, M.P.; Cornberg, M.; Ljunggren, H.G.; Wedemeyer, H.; Bjorkstrom, N.K. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat. Commun. 2018, 9, 2275. [Google Scholar] [CrossRef] [PubMed]
- De Groen, R.A.; Groothuismink, Z.M.A.; van Oord, G.; Kootstra, N.A.; Janssen, H.L.A.; Prins, M.; Schinkel, J.; Boonstra, A. NK cells in self-limited HCV infection exhibit a more extensively differentiated, but not memory-like, repertoire. J. Viral Hepat. 2017, 24, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Collister, M.; Ellison, C.; Li, Q.; Minuk, G.Y.; Rempel, J.D.; Kung, S.K. The Influence of Hepatitis C Viral Loads on Natural Killer Cell Function. Gastroenterol. Res. 2019, 12, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Madrigal-Estebas, L.; McGrath, E.; Conroy, M.J.; Ryan, E.J.; Hegarty, J.E.; O’Farrelly, C.; Doherty, D.G. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut 2008, 57, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Tatsumi, T.; Nishio, A.; Kegasawa, T.; Yoshioka, T.; Yamada, R.; Furuta, K.; Kodama, T.; Shigekawa, M.; Hikita, H.; et al. CEACAM1 Is Associated with the Suppression of Natural Killer Cell Function in Patients With Chronic Hepatitis C. Hepatol. Commun. 2018, 2, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Crotta, S.; Brazzoili, M.; Piccioli, D.; Valiante, N.; Wack, A. Hepatitis C virions subvert natural killer cell activation to generate a cytokine environment permissive for infection. J. Hepatol. 2010, 52, 183–190. [Google Scholar] [CrossRef]
- Farag, M.M.; Weigand, K.; Encke, J.; Momburg, F. Activation of natural killer cells by hepatitis C virus particles in vitro. Clin. Exp. Immunol. 2011, 165, 352–362. [Google Scholar] [CrossRef]
- Brimacombe, C.L.; Wilson, G.K.; Hubscher, S.G.; McKeating, J.A.; Farquhar, M.J. A role for CD81 and hepatitis C virus in hepatoma mobility. Viruses 2014, 6, 1454–1472. [Google Scholar] [CrossRef]
- Prod’homme, V.; Griffin, C.; Aicheler, R.J.; Wang, E.C.; McSharry, B.P.; Rickards, C.R.; Stanton, R.J.; Borysiewicz, L.K.; Lopez-Botet, M.; Wilkinson, G.W.; et al. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1− NK cells. J. Immunol. 2007, 178, 4473–4481. [Google Scholar] [CrossRef]
- Yu, K.; Davidson, C.L.; Wojtowicz, A.; Lisboa, L.; Wang, T.; Airo, A.M.; Villard, J.; Buratto, J.; Sandalova, T.; Achour, A.; et al. LILRB1 polymorphisms influence posttransplant HCMV susceptibility and ligand interactions. J. Clin. Invest. 2018, 128, 1523–1537. [Google Scholar] [CrossRef]
- Prod’homme, V.; Sugrue, D.M.; Stanton, R.J.; Nomoto, A.; Davies, J.; Rickards, C.R.; Cochrane, D.; Moore, M.; Wilkinson, G.W.; Tomasec, P. Human cytomegalovirus UL141 promotes efficient downregulation of the natural killer cell activating ligand CD112. J. Gen. Virol. 2010, 91, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, L.F.; Smyth, M.J.; Martinet, L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; van den Boomen, D.J.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef] [PubMed]
- Hansasuta, P.; Dong, T.; Thananchai, H.; Weekes, M.; Willberg, C.; Aldemir, H.; Rowland-Jones, S.; Braud, V.M. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur. J. Immunol. 2004, 34, 1673–1679. [Google Scholar] [CrossRef]
- Thananchai, H.; Gillespie, G.; Martin, M.P.; Bashirova, A.; Yawata, N.; Yawata, M.; Easterbrook, P.; McVicar, D.W.; Maenaka, K.; Parham, P.; et al. Cutting Edge: Allele-specific and peptide-dependent interactions between KIR3DL1 and HLA-A and HLA-B. J. Immunol. 2007, 178, 33–37. [Google Scholar] [CrossRef]
- Lunemann, S.; Martrus, G.; Holzemer, A.; Chapel, A.; Ziegler, M.; Korner, C.; Garcia Beltran, W.; Carrington, M.; Wedemeyer, H.; Altfeld, M. Sequence variations in HCV core-derived epitopes alter binding of KIR2DL3 to HLA-C * 03:04 and modulate NK cell function. J. Hepatol. 2016, 65, 252–258. [Google Scholar] [CrossRef]
- Wauquier, N.; Petitdemange, C.; Tarantino, N.; Maucourant, C.; Coomber, M.; Lungay, V.; Bangura, J.; Debre, P.; Vieillard, V. HLA-C-restricted viral epitopes are associated with an escape mechanism from KIR2DL2(+) NK cells in Lassa virus infection. EBioMedicine 2019, 40, 605–613. [Google Scholar] [CrossRef]
- Fadda, L.; O’Connor, G.M.; Kumar, S.; Piechocka-Trocha, A.; Gardiner, C.M.; Carrington, M.; McVicar, D.W.; Altfeld, M. Common HIV-1 peptide variants mediate differential binding of KIR3DL1 to HLA-Bw4 molecules. J. Virol. 2011, 85, 5970–5974. [Google Scholar] [CrossRef]
- Naiyer, M.M.; Cassidy, S.A.; Magri, A.; Cowton, V.; Chen, K.; Mansour, S.; Kranidioti, H.; Mbiribindi, B.; Rettman, P.; Harris, S.; et al. KIR2DS2 recognizes conserved peptides derived from viral helicases in the context of HLA-C. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| KIR | Ligand | Notes |
|---|---|---|
| Inhibitory partnerships | ||
| KIR2DL1 | HLA-C2 | |
| KIR2DL2 | HLA-C1 (major); HLA-C2 (minor) | HLA-C1 ligands carry Asp77; HLA-C2 ligands carry Lys77 |
| KIR2DL3 | HLA-C1 | |
| KIR3DL1 | HLA-Bw4 | HLA-Bw4 epitopes can be further subdivided based on the amino acid at position 80 (Ile or Thr) with impacts on NK cell education |
| Activating partnerships | ||
| KIR3DS1 | HLA-F | KIR3DS1 is known to bind HLA-F, but its impact on education is unknown. |
| KIR2DS1 | HLA-C2 | Individuals homozygous for HLA-C2 exhibit tolerized KIR2DS1+ NK cells |
| KIR2DS2 | HLA-A*11 (weak and peptide dependent) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savoy, S.K.A.; Boudreau, J.E. The Evolutionary Arms Race between Virus and NK Cells: Diversity Enables Population-Level Virus Control. Viruses 2019, 11, 959. https://doi.org/10.3390/v11100959
Savoy SKA, Boudreau JE. The Evolutionary Arms Race between Virus and NK Cells: Diversity Enables Population-Level Virus Control. Viruses. 2019; 11(10):959. https://doi.org/10.3390/v11100959
Chicago/Turabian StyleSavoy, Sarah K. A., and Jeanette E. Boudreau. 2019. "The Evolutionary Arms Race between Virus and NK Cells: Diversity Enables Population-Level Virus Control" Viruses 11, no. 10: 959. https://doi.org/10.3390/v11100959
APA StyleSavoy, S. K. A., & Boudreau, J. E. (2019). The Evolutionary Arms Race between Virus and NK Cells: Diversity Enables Population-Level Virus Control. Viruses, 11(10), 959. https://doi.org/10.3390/v11100959