Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype A Reintroduction into Circulation


Abstract
1. Introduction
2. Materials and Methods
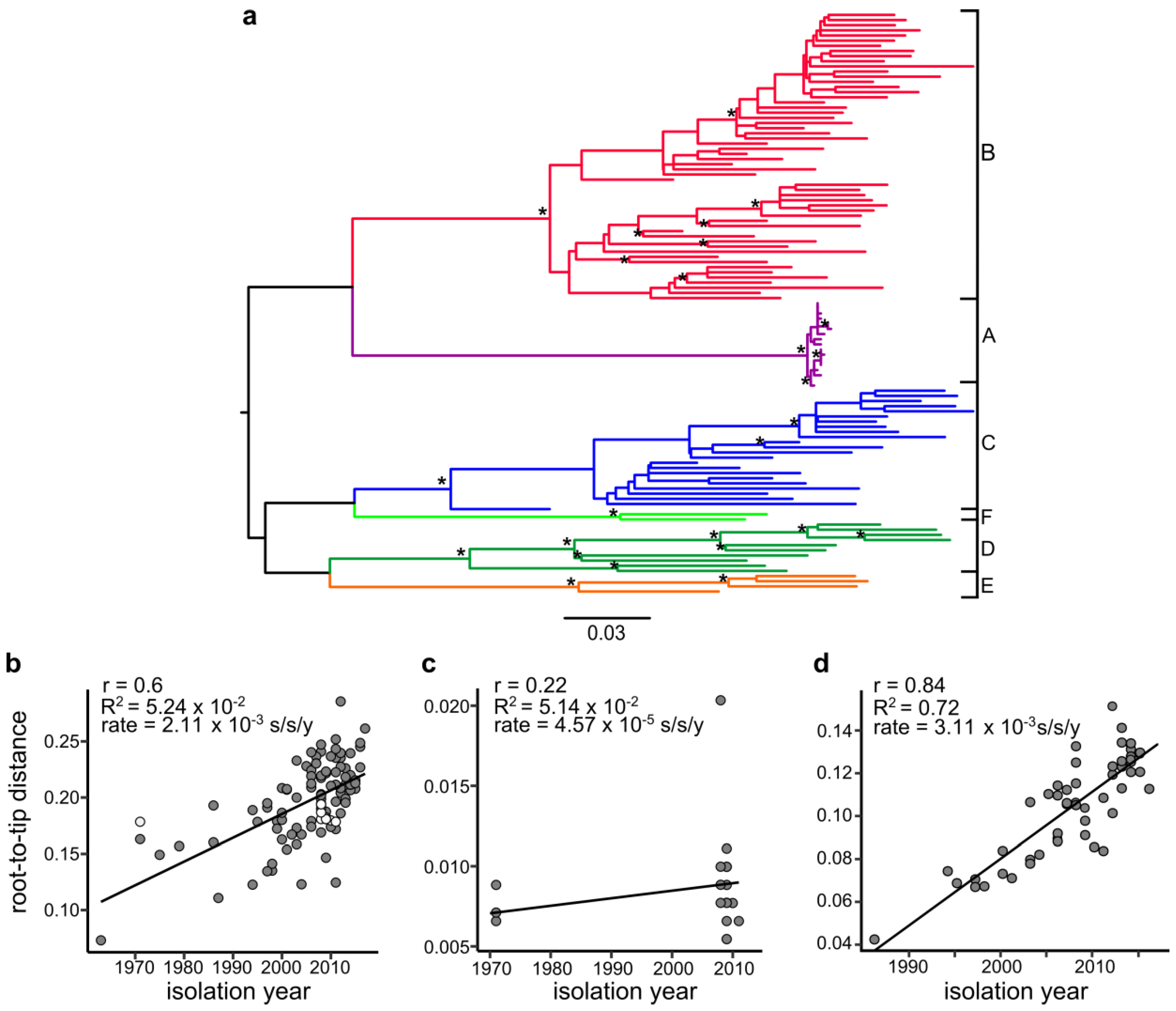
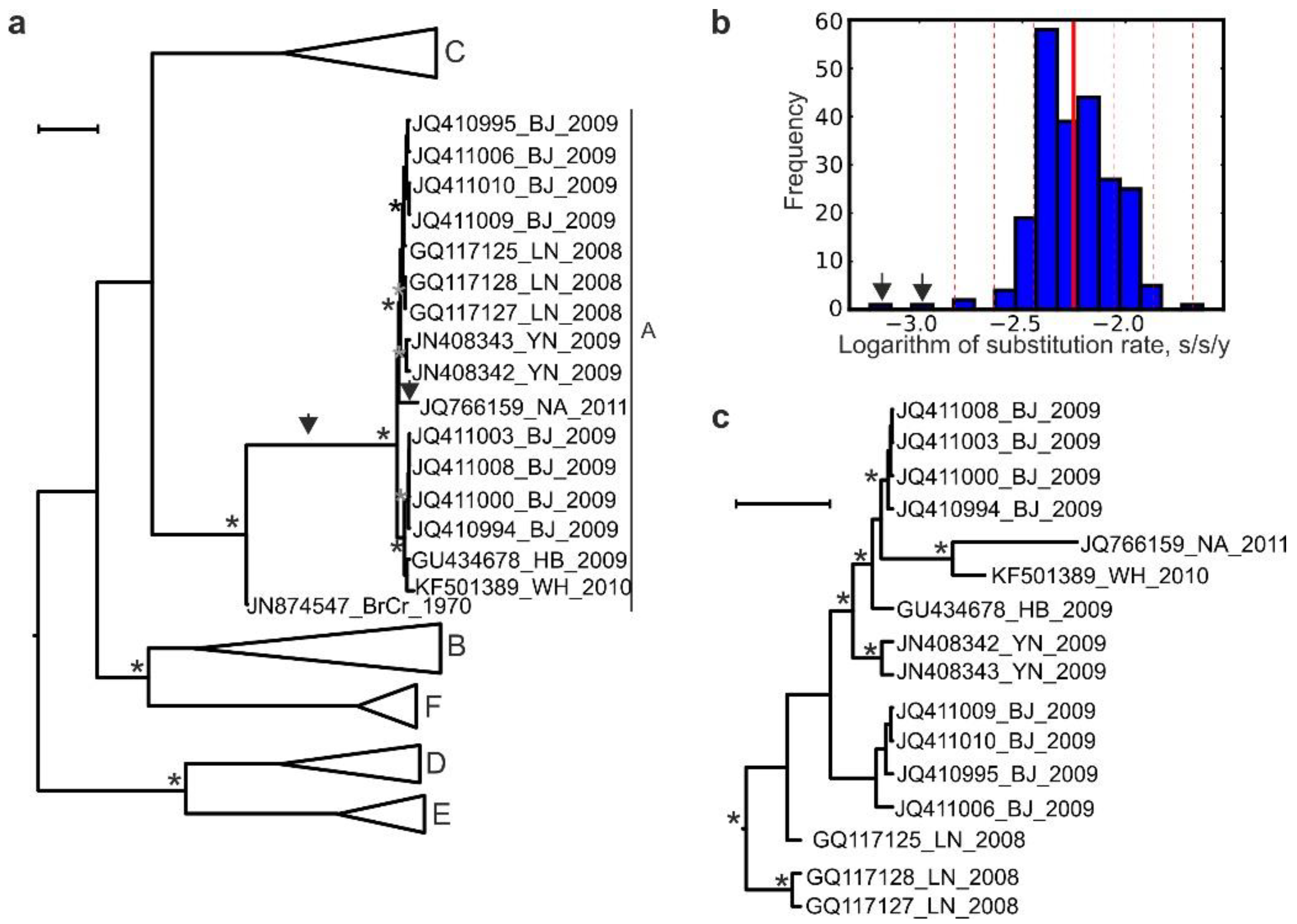
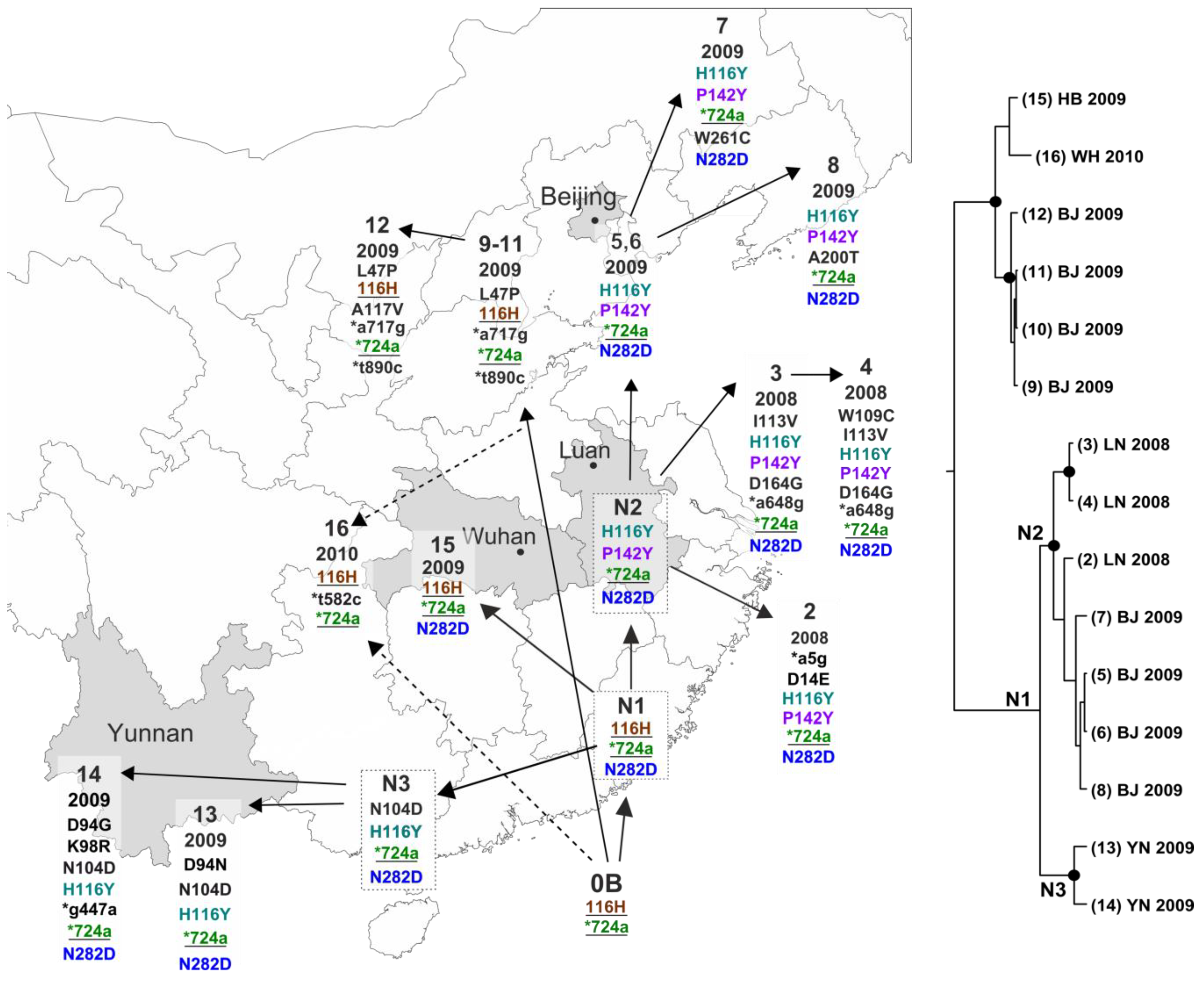
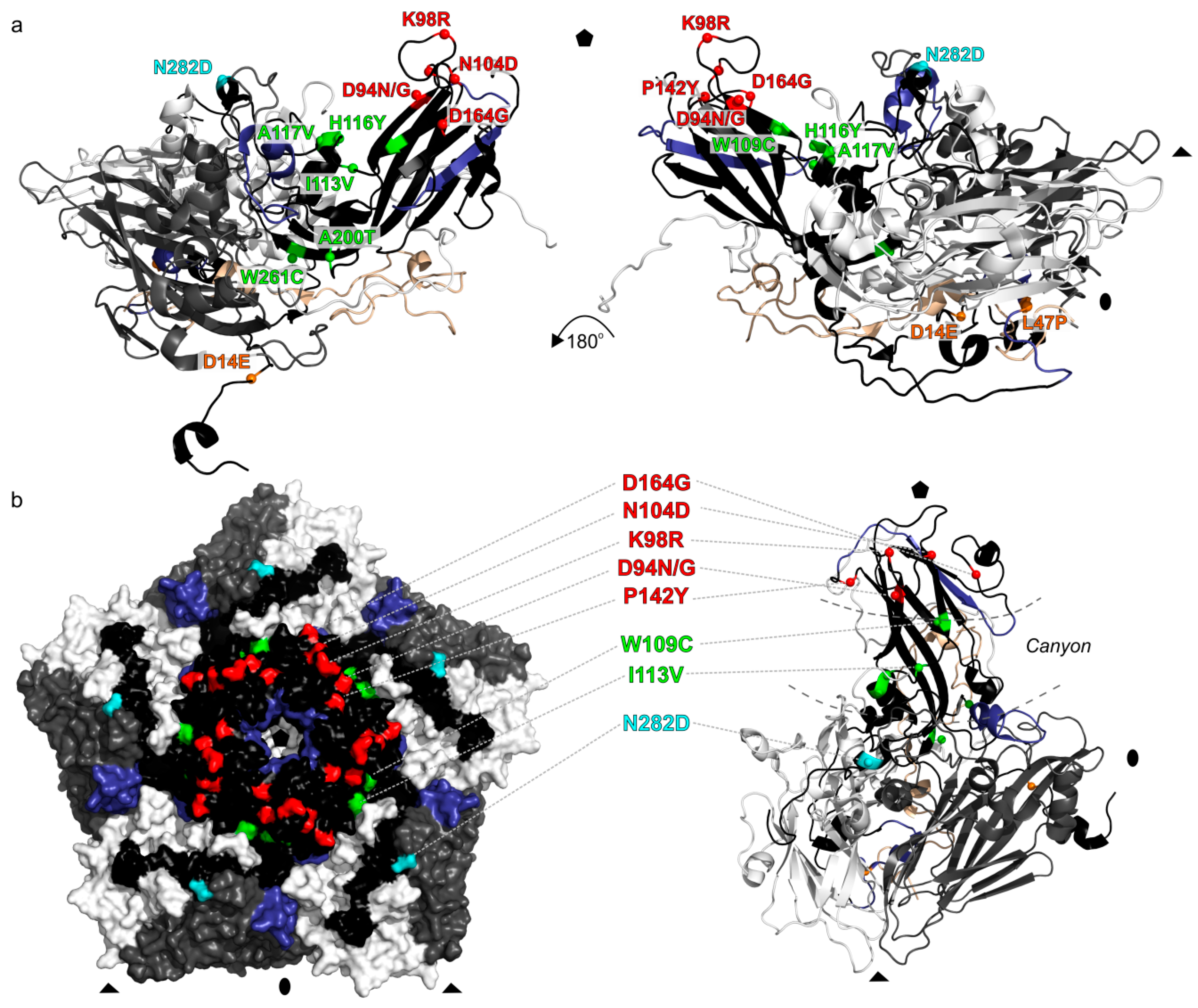
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- McMinn, P.C. Recent advances in the molecular epidemiology and control of human enterovirus 71 infection. Curr. Opin. Virol. 2012, 2, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Tan, X.; Huang, X.; Zhu, S.; Chen, H.; Yu, Q.; Wang, H.; Huo, X.; Zhou, J.; Wu, Y.; Yan, D.; et al. The persistent circulation of enterovirus 71 in people’s republic of China: Causing emerging nationwide epidemics since 2008. PLoS ONE 2011, 6, e25662. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Vakulenko, Y.A.; Turbabina, N.A.; Deviatkin, A.A.; Drexler, J.F. Molecular epidemiology and phylogenetics of human enteroviruses: Is there a forest behind the trees? Rev. Med. Virol. 2018, 28, e2002. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Razafindratsimandresy, R.; Nougairède, A.; Joffret, M.L.; Deshpande, J.M.; Dubot-Pérès, A.; Héraud, J.M.; De Lamballerie, X.; Delpeyroux, F.; Bailly, J.L. Molecular comparison and evolutionary analyses of VP1 nucleotide sequences of new African human enterovirus 71 isolates reveal a wide genetic diversity. PLoS ONE 2014, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, X.J.; Wang, H.Y.; Yan, D.M.; Zhu, S.L.; Wang, D.Y.; Ji, F.; Wang, X.J.; Gao, Y.J.; Chen, L.; et al. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J. Clin. Virol. 2009, 44, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Pillet, S.; Ibrahim, W.; Joffret, M.L.; Pozzetto, B.; Delpeyroux, F.; Gouandjika-Vasilached, I. Molecular characterization of human enteroviruses in the Central African Republic: Uncovering wide diversity and identification of a new human enterovirus A71 genogroup. J. Clin. Microbiol. 2012, 50, 1650–1658. [Google Scholar] [CrossRef]
- Deshpande, J.M.; Nadkarni, S.S.; Francis, P.P. Enterovirus 71 isolated from a case of acute flaccid paralysis in India represents a new genotype. Curr. Sci. 2003, 84, 1350–1353. [Google Scholar]
- Fernandez-Garcia, M.D.; Volle, R.; Joffret, M.L.; Sadeuh-Mba, S.A.; Gouandjika-Vasilache, I.; Kebe, O.; Wiley, M.R.; Majumdar, M.; Simon-Loriere, E.; Sakuntabhai, A.; et al. Genetic characterization of enterovirus A71 circulating in Africa. Emerg. Infect. Dis. 2018, 24, 754–757. [Google Scholar] [CrossRef]
- Schmidt, N.; Lennette, E.; Ho, H. An apparently new enterovirus isolated from patients with disease of the central nervous system. J. Infect. Dis. 1974, 129, 304–309. [Google Scholar] [CrossRef]
- Yang, Z.; Lu, S.; Xian, J.; Ye, J.; Xiao, L.; Luo, J.; Zen, K. Complete genome sequence of a human enterovirus 71 strain isolated in Wuhan, China, in 2010. Genome Announc. 2013, 1, 2012–2013. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, W.; Chang, H.; Tang, R.; Zhao, J.; Gan, L.; Liu, B.; Chen, J.; Wang, M. Genetic analysis of the VP1 region of enterovirus 71 reveals the emergence of genotype A in central China in 2008. Virus Genes 2010, 41, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Luo, Z.; Wang, J.; Xu, Z.; Chen, H.; Fan, D.; Gao, N.; Ping, G.; Zhou, Z.; Zhang, Y.; et al. Phylogenetic Analysis of Enterovirus 71 Circulating in Beijing, China from 2007 to 2009. PLoS ONE 2013, 8, e56318. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.; Carvalho, L.; Pybus, O. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.; Lemey, P.; Baele, G.; Ayres, D.; Drummond, A.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriatesubstitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Lukashev, A.; Vakulenko, Y. Molecular evolution of types in non-polio enteroviruses. J. Gen. Virol 2017, 98, 2968–2981. [Google Scholar] [CrossRef] [PubMed]
- Tracer 1.6. Available online: http://tree.bio.ed.ac.uk/software/tracer (accessed on 1 September 2019).
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 September 2019).
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 42, 95–98. [Google Scholar]
- Tee, K.K.; Lam, T.T.-Y.; Chan, Y.F.; Bible, J.M.; Kamarulzaman, A.; Tong, C.Y.W.; Takebe, Y.; Pybus, O.G. Evolutionary genetics of human enterovirus 71: Origin, population dynamics, natural selection, and seasonal periodicity of the VP1 gene. J. Virol. 2010, 84, 3339–3350. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Shumilina, E.Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, J.F.; Drosten, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95, 868–873. [Google Scholar] [CrossRef] [PubMed]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Koike, S.; Kroes, A.C.M.; Lukashev, A.; Perera, D.; Roivainen, M.; et al. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J. Virol. 2012, 86, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Shimizu, H.; Nagata, N.; Ami, Y.; Suzaki, Y.; Sata, T.; Iwasaki, T.; Miyamura, T. Temperature-sensitive mutants of enterovirus 71 show attenuation in cynomolgus monkeys. J. Gen. Virol. 2005, 86, 1391–1401. [Google Scholar] [CrossRef]
- Huang, M.-L.; Chiang, P.-S.; Chia, M.-Y.; Luo, S.-T.; Chang, L.-Y.; Lin, T.-Y.; Ho, M.-S.; Lee, M.-S. Cross-reactive neutralizing antibody responses to enterovirus 71 infections in young children: Implications for vaccine development. PLoS Negl. Trop. Dis. 2013, 7, e2067. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G.; He, Y.; Kuhn, R.J. Picornavirus-receptor interactions. Trends Microbiol. 2002, 10, 324–331. [Google Scholar] [CrossRef]
- He, Y.; Bowman, V.D.; Mueller, S.; Bator, C.M.; Bella, J.; Peng, X.; Baker, T.S.; Wimmer, E.; Kuhn, R.J.; Rossmann, M.G. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA 2000, 97, 79–84. [Google Scholar] [CrossRef]
- He, Y.; Mueller, S.; Chipman, P.R.; Bator, C.M.; Peng, X.; Bowman, V.D.; Mukhopadhyay, S.; Wimmer, E.; Kuhn, R.J.; Rossmann, M.G. Complexes of poliovirus serotypes with their common cellular receptor, CD155. J. Virol. 2003, 77, 4827–4835. [Google Scholar] [CrossRef]
- Olson, N.H.; Kolatkar, P.R.; Oliveira, M.A.; Cheng, R.H.; Grevet, J.M.; McClelland, A.; Baker, T.S.; Rossmann, M.G. Structure of a human rhinovirus complexed with its receptor molecule. Proc. Natl. Acad. Sci. USA 1993, 90, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Shen, L.; Wu, J.; Zou, X.; Gu, J.; Chen, J.; Mao, L. Enterovirus A71 proteins: Structure and function. Front. Microbiol. 2018, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.Y.A.; Chen, M.F.; Huang, Y.C.; Shih, S.R.; Chiu, C.H.; Lin, J.J.; Wang, J.R.; Tsao, K.C.; Lin, T.Y. Epitope-associated and specificity-focused features of EV71-neutralizing antibody repertoires from plasmablasts of infected children. Nat. Commun. 2017, 8, 762. [Google Scholar] [CrossRef] [PubMed]
- De Colibus, L.; Wang, X.; Spyrou, J.A.B.; Kelly, J.; Ren, J.; Grimes, J.; Puerstinger, G.; Stonehouse, N.; Walter, T.S.; Hu, Z.; et al. More-powerful virus inhibitors from structure-based analysis of Hev71 capsid-binding molecules. Nat. Struct. Mol. Biol. 2014, 21, 282–288. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Brault, A.C.; Powers, A.M.; Medina, G.; Wang, E.; Kang, W.; Salas, R.A.; De Siger, J.; Weaver, S.C. Potential sources of the 1995 venezuelan equine encephalitis subtype IC epidemic. J. Virol. 2001, 75, 5823–5832. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O. The Re-Emergence of H1N1 influenza virus in 1977: A cautionary tale for estimating divergence times using biologically unrealistic sampling dates. PLoS ONE 2010, 5, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, Y.A.; Deviatkin, A.A.; Lukashev, A.N. The effect of sample bias and experimental artefacts on statistical phylogenetic analysis of picornaviruses. Viruses 2019. submitted. [Google Scholar]
- Famulare, M.; Chang, S.; Iber, J.; Zhao, K.; Adeniji, J.A.; Bukbuk, D.; Baba, M. Sabin Vaccine reversion in the field: A comprehensive analysis of Sabin-like poliovirus isolates in Nigeria. J. Virol. 2016, 90, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Hassel, C.; Mirand, A.; Lukashev, A.; Terletskaialadwig, E.; Farkas, A.; Schuffenecker, I.; Diedrich, S.; Huemer, H.P.; Bernard, U.C.; Virpath, E.A. Transmission patterns of human enterovirus 71 to, from and among European countries, 2003 to 2013. Euro. Surveill. 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Koroleva, G.A.; Karmysheva, V.Y.; Lukashev, A.N. Enterovirus 71 pathogenicity in monkeys and cotton rats. Arch. Virol. 2013, 159, 1133–1138. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Bendig, J.; Cabrerizo, M.; Cardosa, J.; Hyypia, T.; Ivanova, O.E.; Kelly, A.; Kroes, A.C.M.; Lukashev, A.; MacAdam, A.; et al. Transmission networks and population turnover of echovirus 30. J. Virol. 2009, 83, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Desselberger, U.; Palese, P. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature 1978, 274, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Shooter, R.A. Report of the Investigation into the Cause of the 1978 Birmingham Smallpox Occurrence; Department of Health, Social Services and Public Safety: London, UK, 1980. [Google Scholar]
- Furmanski, M. Threatened Pandemics and Laboratory Escapes: Self-Fulfilling Prophecies. Available online: https://thebulletin.org/2014/03/threatened-pandemics-and-laboratory-escapes-self-fulfilling-prophecies/ (accessed on 10 July 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Figure 3 ID | GenBank Accession | Location | Year | Reference | Synonymous Nucleotide Substitutions | Amino Acid Substitutions | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 447 | 582 | 648 | 717 | 724 | 890 | 14 | 47 | 94 | 98 | 104 | 109 | 113 | 116 | 117 | 142 | 164 | 200 | 261 | 282 | |||||
| 0A | AB204853 | * | - | [28] | a | g | t | a | a | c | t | D | L | D | K | N | W | I | H | A | P | D | A | W | N |
| 1 | JQ766159 | Unknown | 2011 | D.s. | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 0B | JN874547 | the USA | 1970 | [29] | a | g | t | a | a | a | t | D | L | D | K | N | W | I | H | A | P | D | A | W | N |
| N1 ** | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | D |
| N2 ** | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Y | - | - | - | D |
| 2 | GQ117125 | Luan | 2008 | [12] | g | - | - | - | - | - | - | E | - | - | - | - | - | - | Y | - | Y | - | - | - | D |
| 3 | GQ117128 | Luan | 2008 | [12] | - | - | - | g | - | - | - | - | - | - | - | - | - | V | Y | - | Y | G | - | - | D |
| 4 | GQ117127 | Luan | 2008 | [12] | - | - | - | g | - | - | - | - | - | - | - | - | C | V | Y | - | Y | G | - | - | D |
| 5 | JQ411010 | Beijing | 2009 | [13] | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Y | - | Y | - | - | - | D |
| 6 | JQ411009 | Beijing | 2009 | [13] | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Y | - | Y | - | - | - | D |
| 7 | JQ411006 | Beijing | 2009 | [13] | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Y | - | Y | - | - | C | D |
| 8 | JQ410995 | Beijing | 2009 | [13] | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Y | - | Y | - | T | - | D |
| 9 | JQ411003 | Beijing | 2009 | [13] | - | - | - | - | g | - | c | - | P | - | - | - | - | - | - | - | - | - | - | - | - |
| 10 | JQ411000 | Beijing | 2009 | [13] | - | - | - | - | g | - | c | - | P | - | - | - | - | - | - | - | - | - | - | - | - |
| 11 | JQ411008 | Beijing | 2009 | [13] | - | - | - | - | g | - | c | - | P | - | - | - | - | - | - | - | - | - | - | - | - |
| 12 | JQ410994 | Beijing | 2009 | [13] | - | - | - | - | g | - | c | - | P | - | - | - | - | - | - | V | - | - | - | - | - |
| 15 | GU434678 | Hubei province | 2009 | D.s | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | D |
| 16 | KF501389 | Wuhan | 2010 | [11] | - | - | c | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| N3 ** | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Y | - | - | - | - | - | D |
| 13 | JN408342 | Yunnan | 2009 | D.s. | - | - | - | - | - | - | - | - | - | N | - | D | - | - | Y | - | - | - | - | - | D |
| 14 | JN408343 | Yunnan | 2009 | D.s. | - | a | - | - | - | - | - | - | - | G | R | D | - | - | Y | - | - | - | - | - | D |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakulenko, Y.; Deviatkin, A.; Lukashev, A. Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype A Reintroduction into Circulation. Viruses 2019, 11, 895. https://doi.org/10.3390/v11100895
Vakulenko Y, Deviatkin A, Lukashev A. Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype A Reintroduction into Circulation. Viruses. 2019; 11(10):895. https://doi.org/10.3390/v11100895
Chicago/Turabian StyleVakulenko, Yulia, Andrei Deviatkin, and Alexander Lukashev. 2019. "Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype A Reintroduction into Circulation" Viruses 11, no. 10: 895. https://doi.org/10.3390/v11100895
APA StyleVakulenko, Y., Deviatkin, A., & Lukashev, A. (2019). Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype A Reintroduction into Circulation. Viruses, 11(10), 895. https://doi.org/10.3390/v11100895