The Robust Self-Assembling Tubular Nanostructures Formed by gp053 from Phage vB_EcoM_FV3



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning Procedures
2.2. Protein Expression
2.3. Protein Purification
2.4. Limited Proteolysis of Recombinant Proteins with Trypsin
2.5. In-Gel Protein Digestion for Mass Spectrometry Analysis, Liquid Chromatography and Mass Spectrometry
2.6. Labelling of gp053 Polysheaths with Neutravidin-Conjugated Gold Nanoparticles
2.7. Transmission Electron Microscopy (TEM)
2.8. Bioinformatics and Molecular Modeling
3. Results
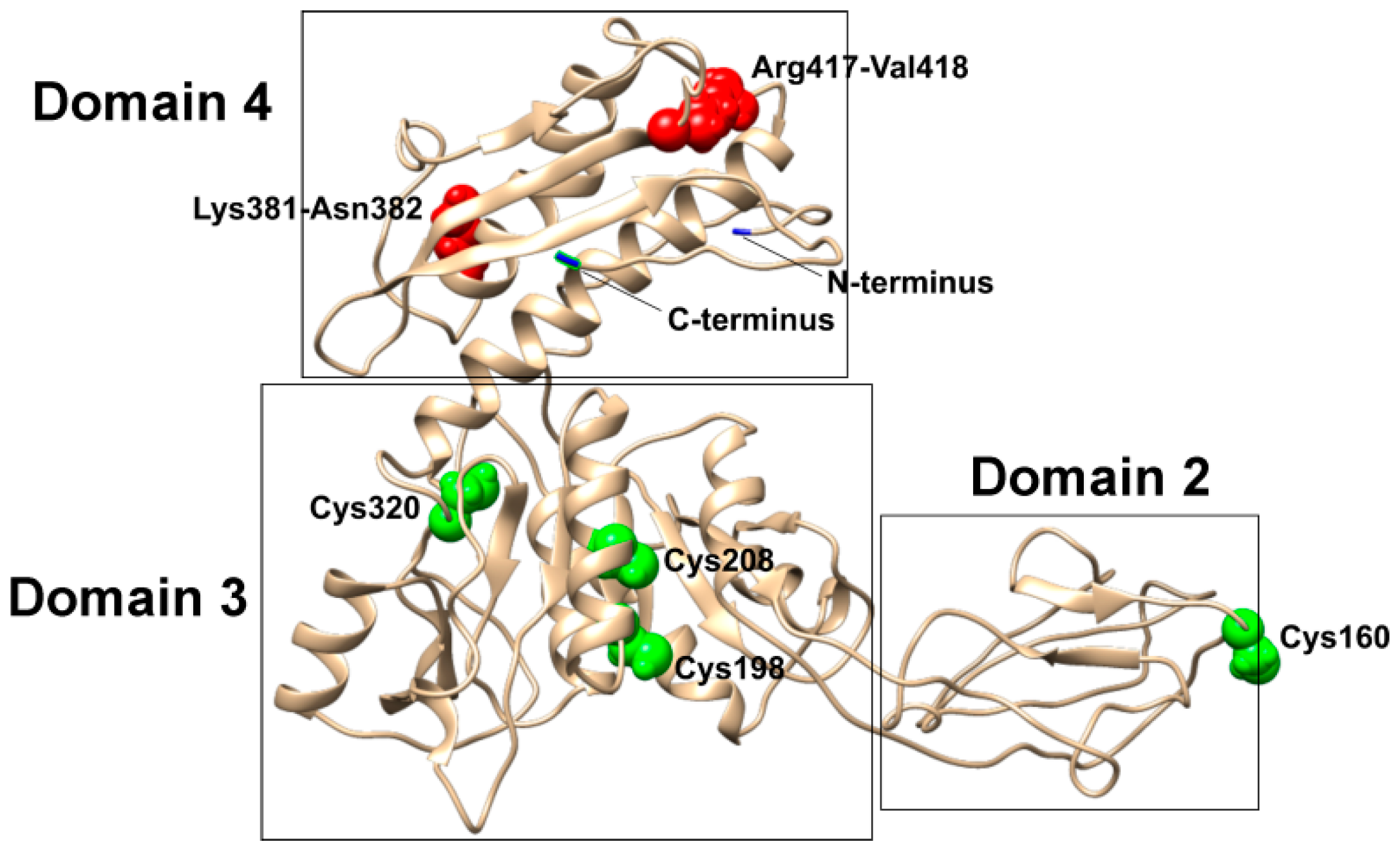
3.1. Bioinformatics Analysis
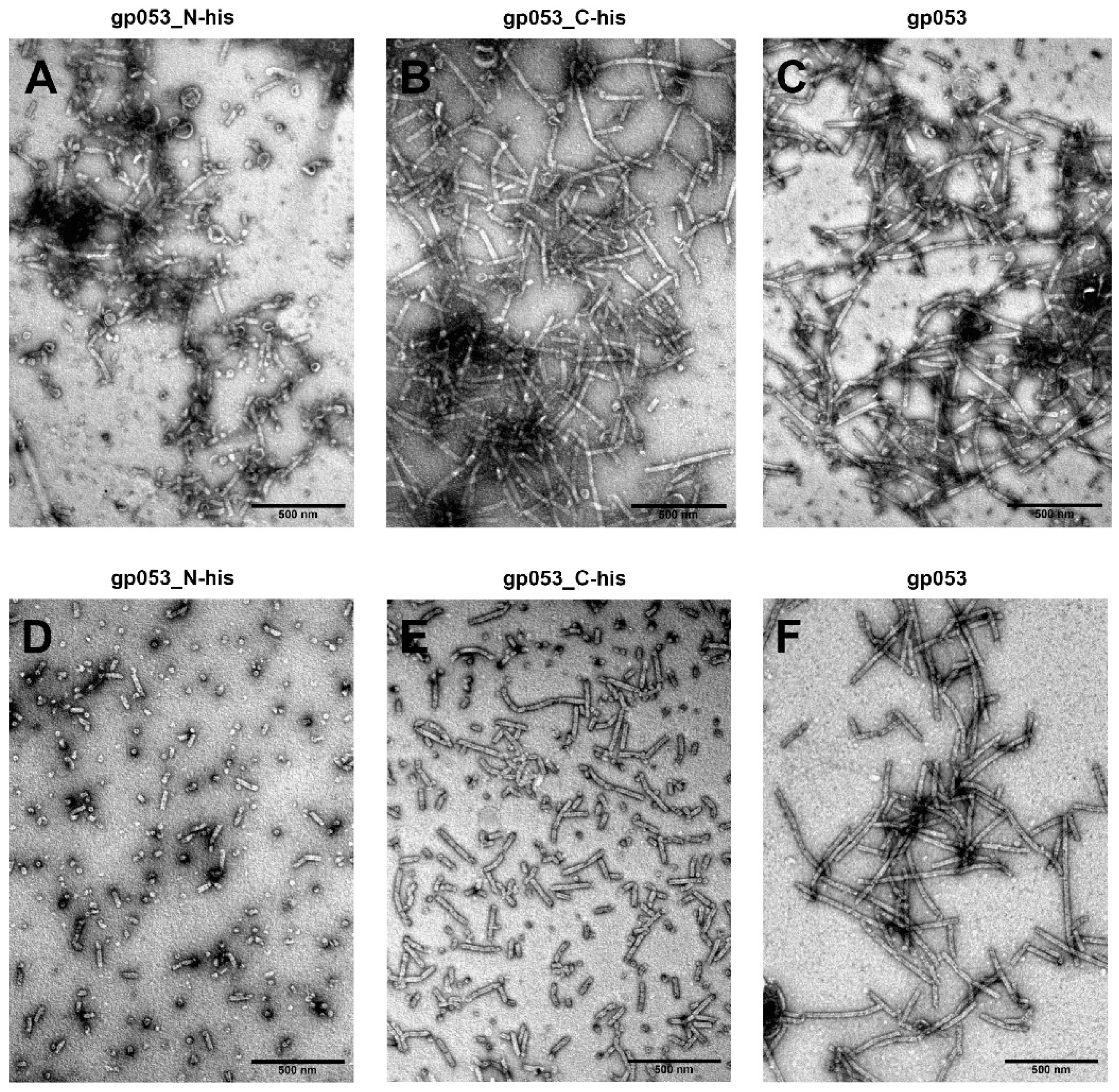
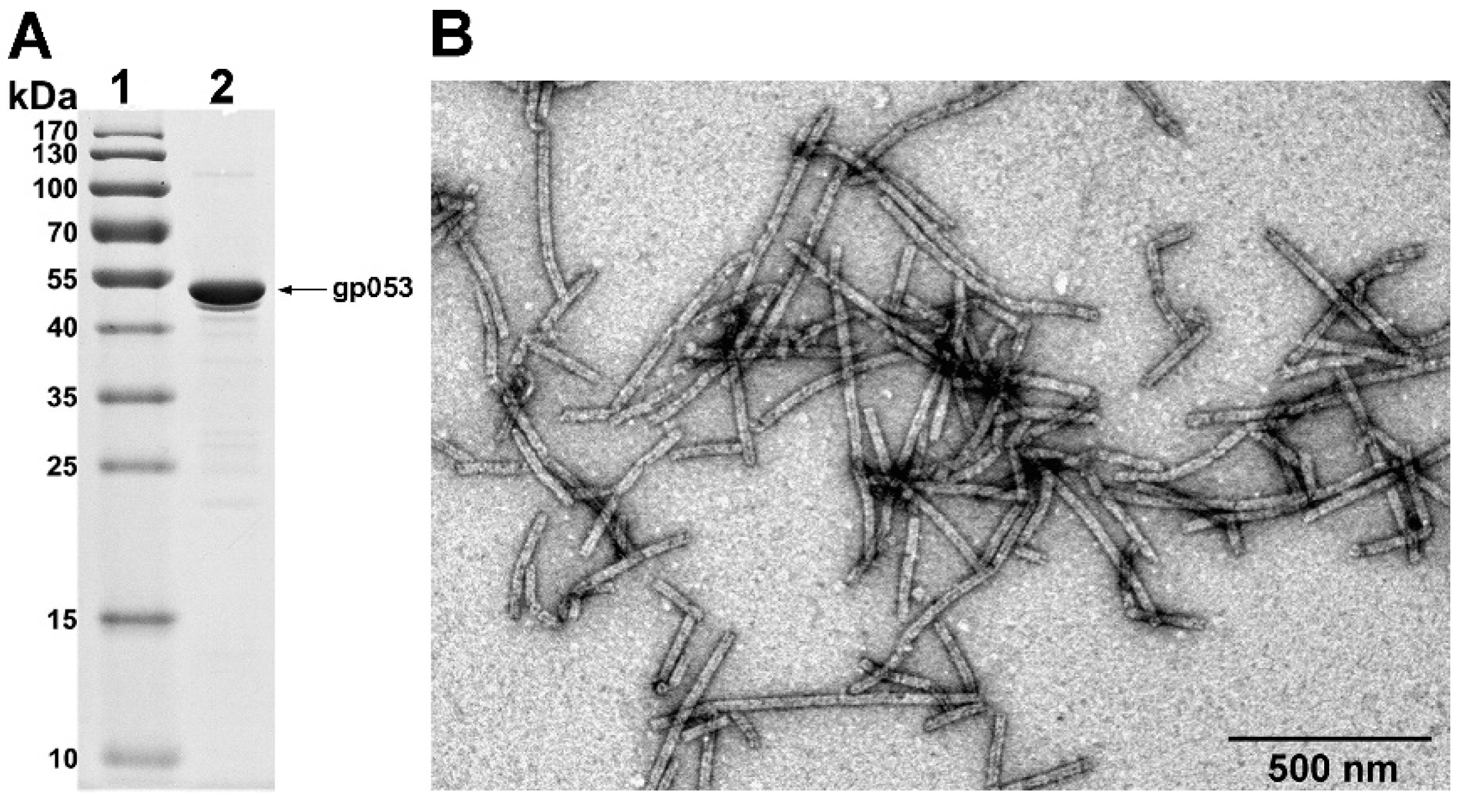
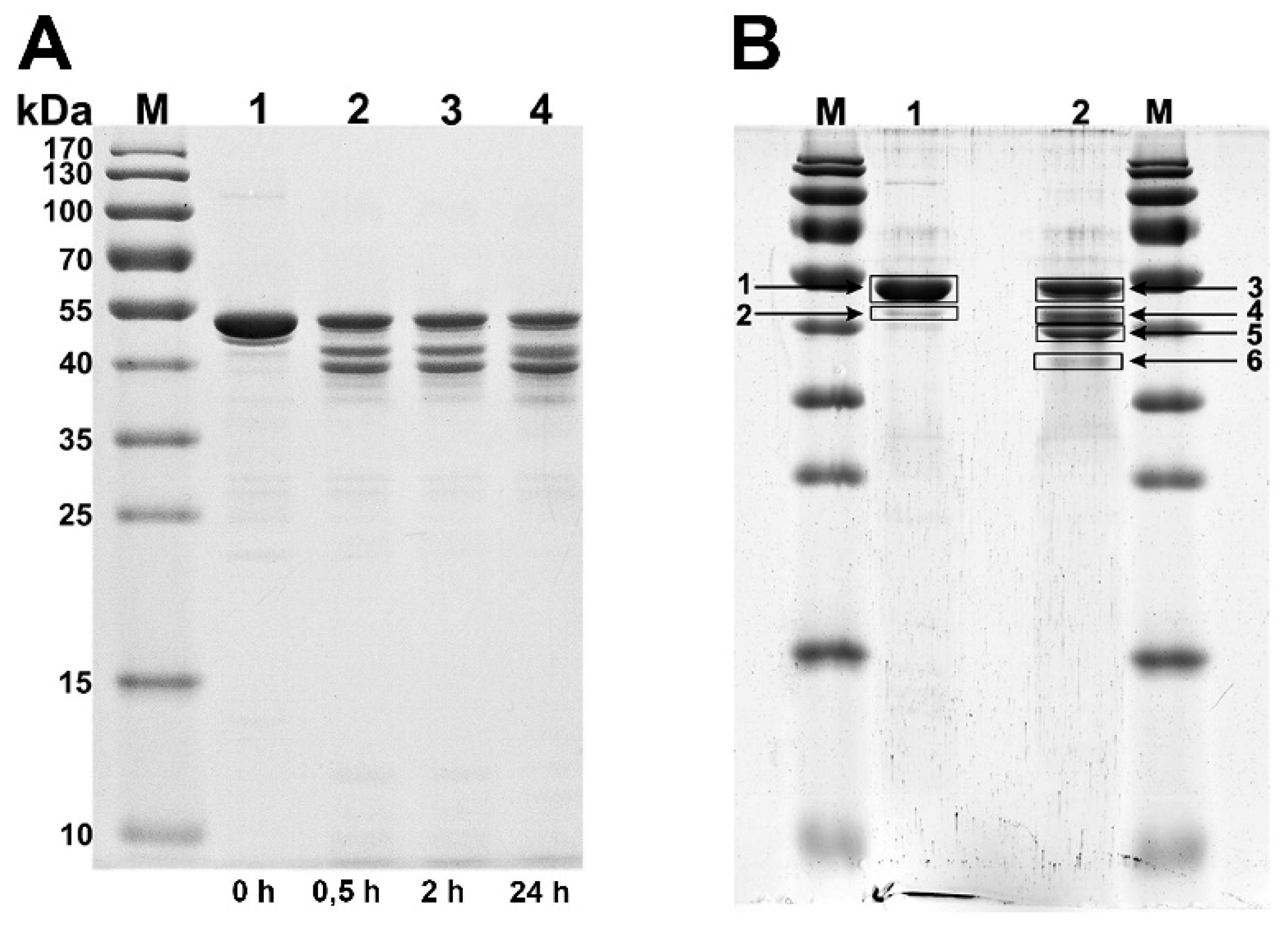
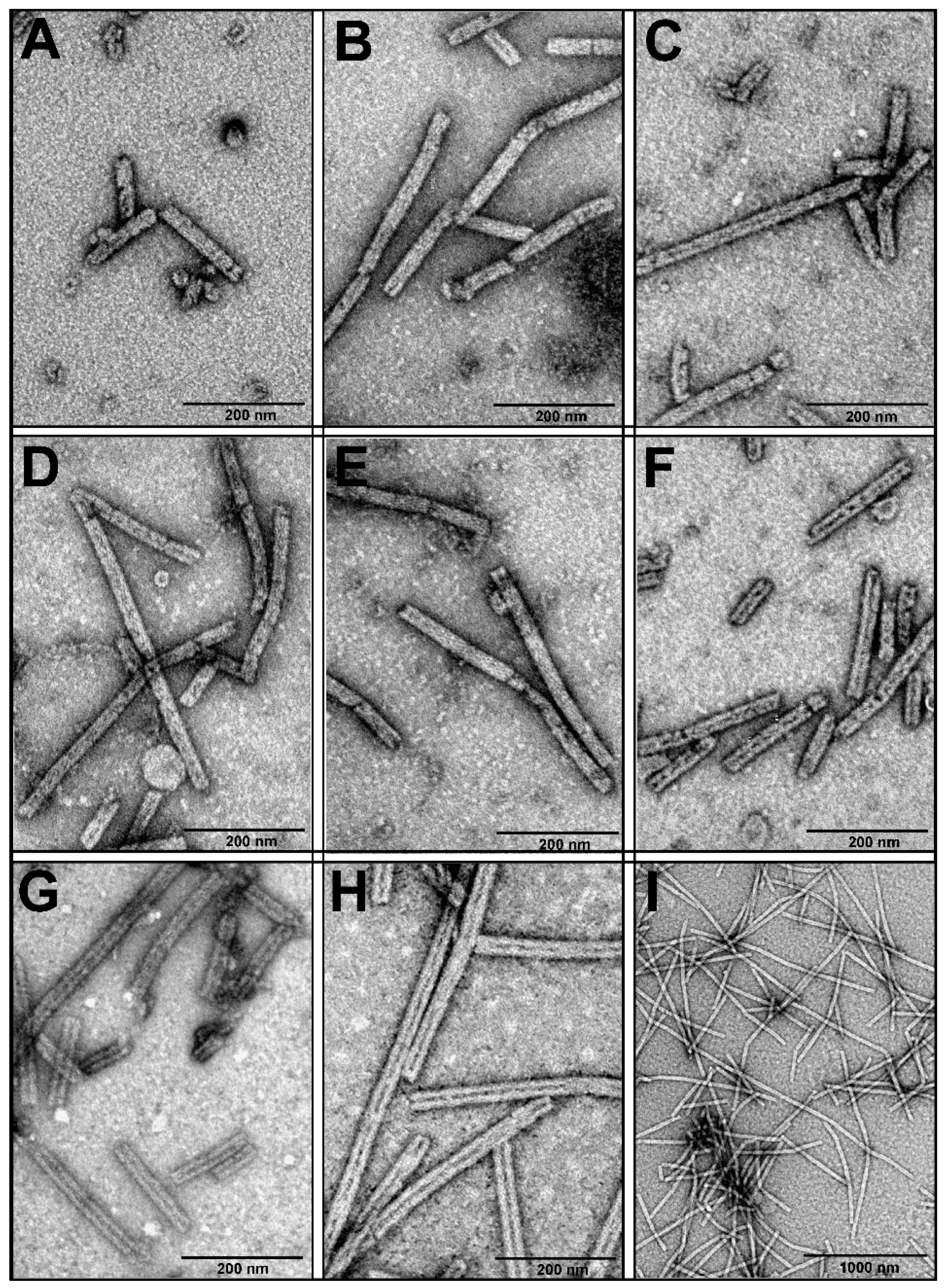
3.2. Production and Analysis of the Recombinant gp053
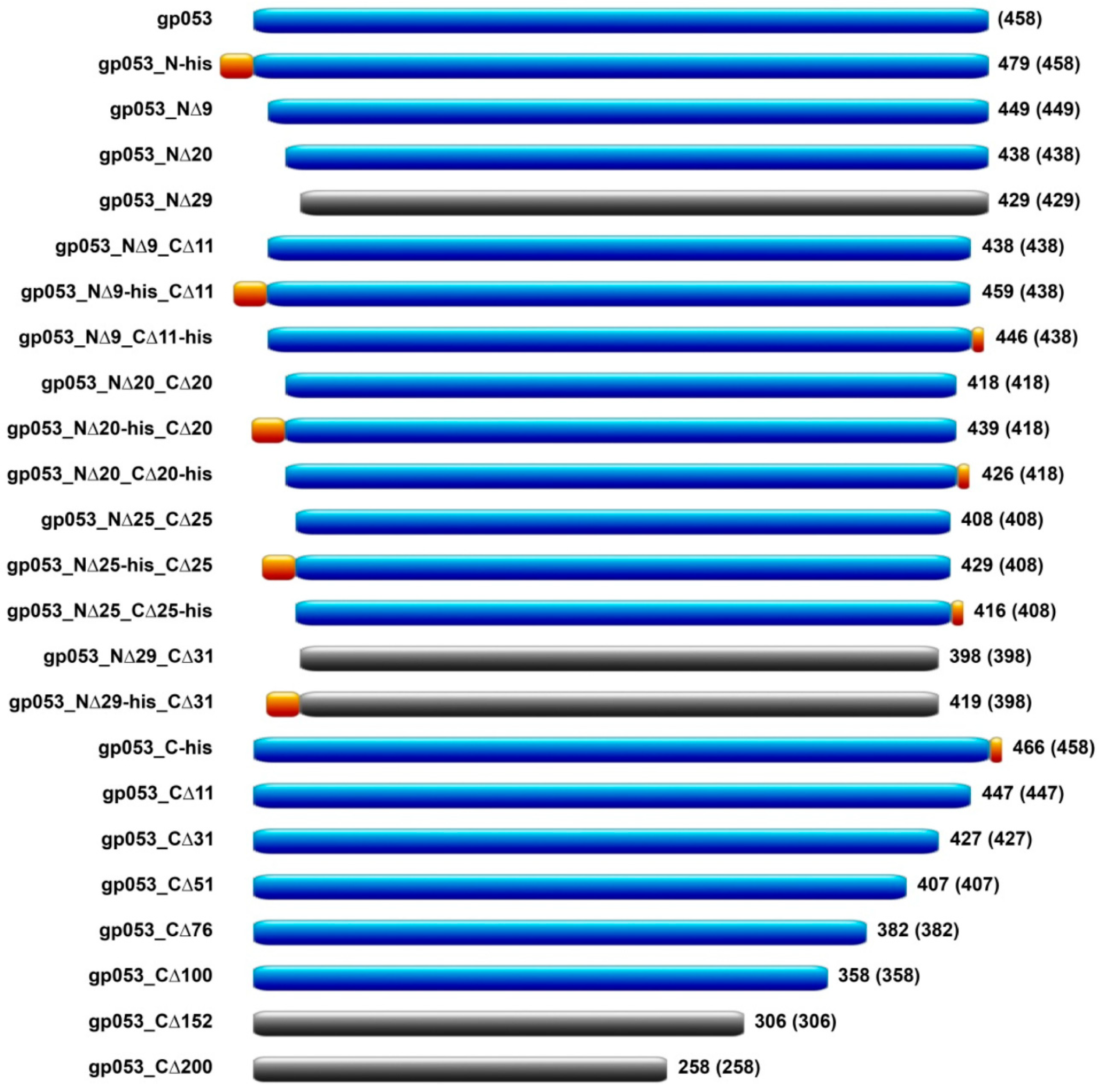
3.3. Construction of gp053 Mutants
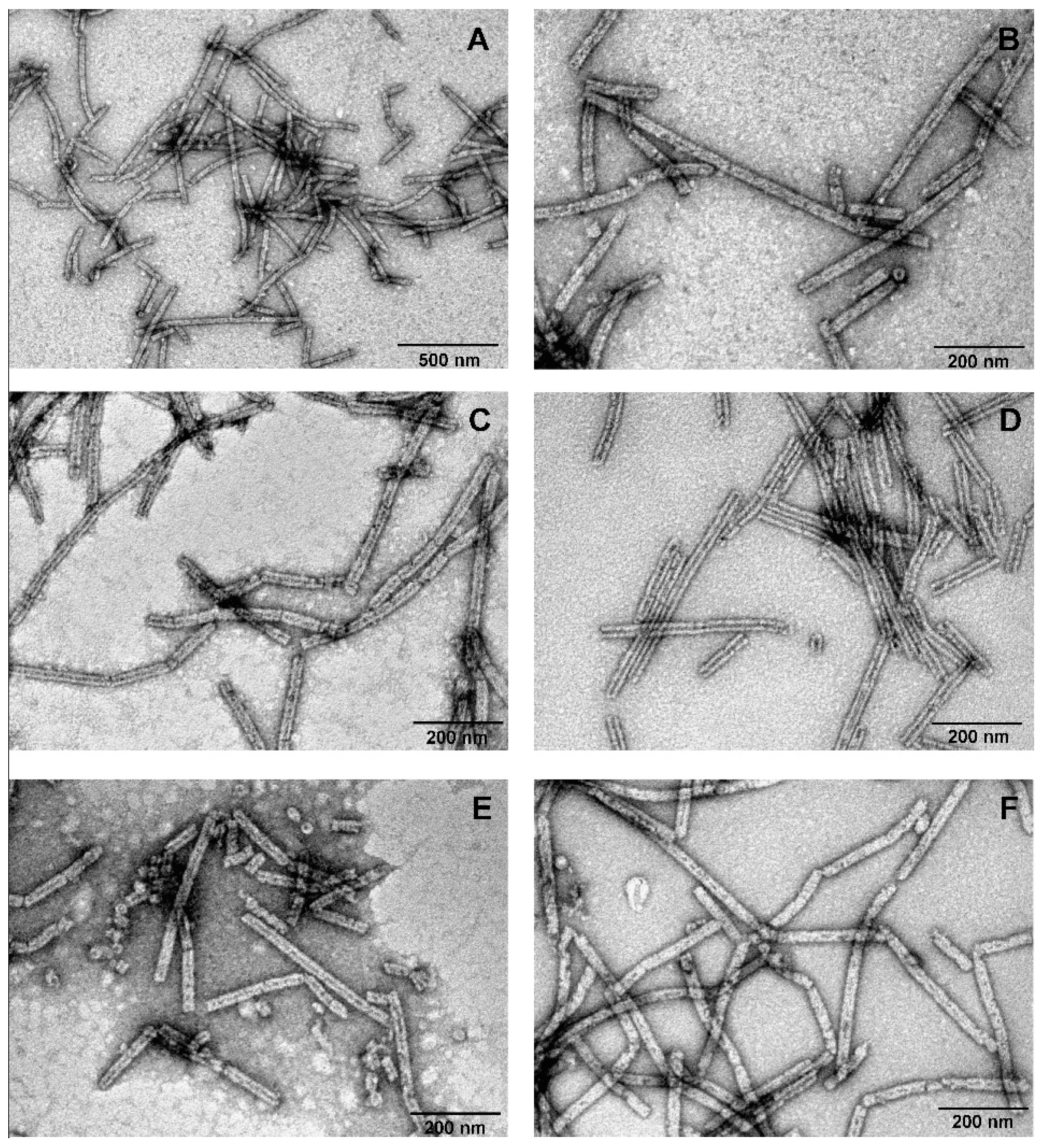
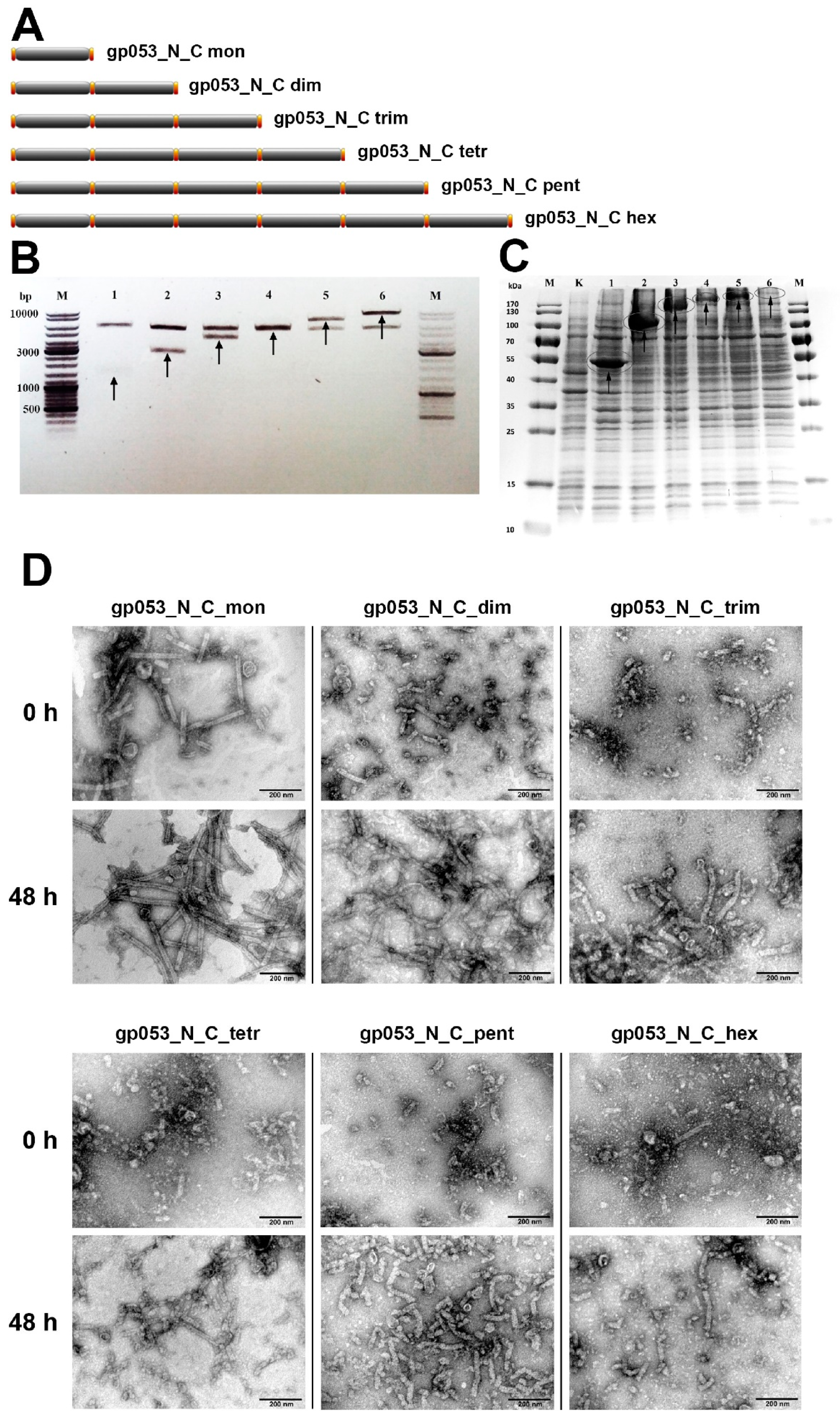
3.4. Investigation of the Oligomeric Constructs of gp053
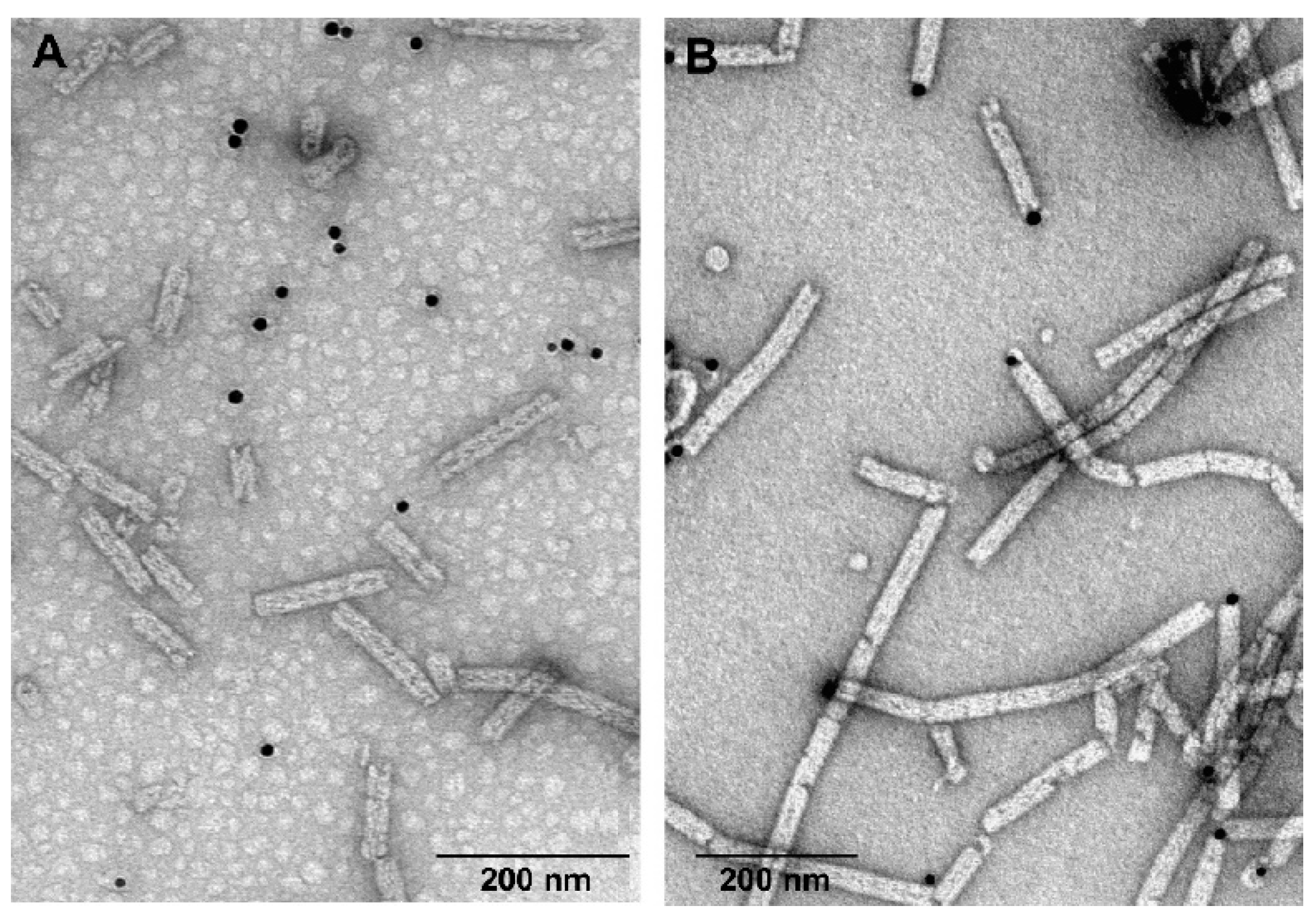
3.5. Labelling of gp053 Polysheaths with Neutravidin-Conjugated Gold Nanoparticles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Busseron, E.; Ruff, Y.; Moulin, E.; Giuseppone, N. Supramolecular self-assemblies as functional nanomaterials. Nanoscale 2013, 5, 7098–7140. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Q. Fabrication of nanoarchitectures templated by virus-based nanoparticles: Strategies and applications. Small 2014, 10, 230–245. [Google Scholar] [CrossRef]
- Lee, E.J.; Lee, N.K.; Kim, I.S. Bioengineered protein-based nanocage for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N.; Inaba, H.; Terauchi, M.; Stieg, A.Z.; Sanghamitra, N.J.; Koshiyama, T.; Yutani, K.; Kanamaru, S.; Arisaka, F.; Hikage, T.; et al. Construction of robust bio-nanotubes using the controlled self-assembly of component proteins of bacteriophage T4. Small 2010, 6, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.C.; Soto, C.M.; Chen, M.S.; Bruckman, M.A.; Moore, M.H.; Barry, E.; Ratna, B.R.; Pehrsson, P.E.; Spies, B.R.; Confer, T.S. Biotemplating rod-like viruses for the synthesis of copper nanorods and nanowires. J. Nanobiotechnol. 2012, 10, 18. [Google Scholar] [CrossRef]
- Drygin, Y.; Kondakova, O.; Atabekov, J. Production of platinum atom nanoclusters at one end of helical plant viruses. Adv. Virol. 2013, 2013, 746796. [Google Scholar] [CrossRef]
- Swaminathan, S.; Cui, Y. Biochemical functionalization of peptide nanotubes with phage displayed peptides. Nanotechnology 2016, 27, 365703. [Google Scholar] [CrossRef]
- Huang, Y.; Chiang, C.Y.; Lee, S.K.; Gao, Y.; Hu, E.L.; de Yoreo, J.; Belcher, A.M. Programmable assembly of nanoarchitectures using genetically engineered viruses. Nano Lett. 2005, 5, 1429–1434. [Google Scholar] [CrossRef]
- Singh, P.; Gonzalez, M.J.; Manchester, M. Viruses and their uses in nanotechnology. Drug Dev. Res. 2006, 67, 23–41. [Google Scholar] [CrossRef]
- Young, M.; Willits, D.; Uchida, M.; Douglas, T. Plant viruses as biotemplates for materials and their use in nanotechnology. Annu. Rev. Phytopathol. 2008, 46, 361–384. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lim, J.S.; Harris, M.T. Synthesis and application of virus-based hybrid nanomaterials. Biotechnol. Bioeng. 2012, 109, 16–30. [Google Scholar] [CrossRef]
- Hyman, P. Bacteriophages and nanostructured materials. Adv. Appl. Microbiol. 2012, 78, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Molek, P.; Bratkovič, T. Bacteriophages as scaffolds for bipartite display: Designing swiss army knives on a nanoscale. Bioconjug. Chem. 2015, 3, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically engineered phages: A review of advances over the last decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Ju, Z.; Sun, W. Drug delivery vectors based on filamentous bacteriophages and phage-mimetic nanoparticles. Drug Deliv. 2017, 24, 1898–1908. [Google Scholar] [CrossRef]
- Kim, I.; Moon, J.S.; Oh, J.W. Recent advances in M13 bacteriophage-based optical sensing applications. Nano Converg. 2016, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Fan, B.; Samdin, T.D.; Monteiro, D.A.; Desai, M.S.; Scheideler, O.; Jin, H.E.; Kim, S.; Lee, S.W. Phage-based structural color sensors and their pattern recognition sensing system. ACS Nano 2017, 11, 3632–3641. [Google Scholar] [CrossRef]
- Lee, S.W.; Belcher, A.M. Virus-based fabrication of micro- and nanofibers using electrospinning. Nano Lett. 2004, 4, 387–390. [Google Scholar] [CrossRef]
- Merzlyak, A.; Indrakanti, S.; Lee, S.W. Genetically engineered nanofiber-like viruses for tissue regenerating materials. Nano Lett. 2009, 9, 846–852. [Google Scholar] [CrossRef]
- Yang, S.H.; Chung, W.J.; McFarland, S.; Lee, S.W. Assembly of bacteriophage into functional materials. Chem. Rec. 2013, 13, 43–59. [Google Scholar] [CrossRef]
- Lee, J.H.; Warner, C.M.; Jin, H.E.; Barnes, E.; Poda, A.R.; Perkins, E.J.; Lee, S.W. Production of tunable nanomaterials using hierarchically assembled bacteriophages. Nat. Protoc. 2017, 12, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, V.; Han, J.; Kim, C.; Kang, Y.C.; Oh, J.W. Self-assembled nanoporous biofilms from functionalized nanofibrous M13 bacteriophage. Viruses 2018, 10, 322. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Serizawa, T. Filamentous viruses as building blocks for hierarchical self-assembly toward functional soft materials. Bull. Chem. Soc. Jpn. 2018, 91, 455–466. [Google Scholar] [CrossRef]
- Nam, K.T.; Peelle, B.R.; Lee, S.W.; Belcher, A.M. Genetically driven assembly of nanorings based on the M13 virus. Nano Lett. 2004, 4, 23–27. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Walker-Kopp, N.; Wilkens, S.; Cingolani, G. Foldon-guided self-assembly of ultra-stable protein fibers. Protein Sci. 2008, 17, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Guo, P. Bacterial virus phi29 DNA-packaging motor and its potential applications in gene therapy and nanotechnology. Methods Mol. Biol. 2005, 300, 285–324. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Valluzzi, R.; Goldberg, E. Design of protein struts for self-assembling nanoconstructs. Proc. Natl. Acad. Sci. USA 2002, 99, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- Daube, S.S.; Arad, T.; Bar-Ziv, R. Cell-free co-synthesis of protein nanoassemblies: Tubes, rings, and doughnuts. Nano Lett. 2007, 7, 638–641. [Google Scholar] [CrossRef]
- Aksyuk, A.A.; Leiman, P.G.; Kurochkina, L.P.; Shneider, M.M.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail sheath structure of bacteriophage T4: A molecular machine for infecting bacteria. EMBO J. 2009, 28, 821–829. [Google Scholar] [CrossRef]
- Arisaka, F.; Yap, M.L.; Kanamaru, S.; Rossmann, M.G. Molecular assembly and structure of the bacteriophage T4 tail. Biophys. Rev. 2016, 8, 385–396. [Google Scholar] [CrossRef]
- Fokine, A.; Rossmann, M.G. Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Shneider, M.M. Contractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Chipman, P.R.; Leiman, P.G.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 2005, 12, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Boy de la Tour, E. On the fine structure of normal and ‘‘polymerized’’ tail sheath of phage T4. J. Ultrastruct. Res. 1964, 11, 545–563. [Google Scholar] [CrossRef]
- Moody, M.F. Structure of the sheath of bacteriphage T4. I. Structure of the contracted sheath and polysheath. J. Mol. Biol. 1967, 25, 167–200. [Google Scholar] [CrossRef]
- Tschopp, J.; Arisaka, F.; van Driel, R.; Engel, J. Purification, characterization and reassembly of the bacteriophage T4D tail sheath protein P18. J. Mol. Biol. 1979, 128, 247–258. [Google Scholar] [CrossRef]
- Kurochkina, L.P.; Aksyuk, A.A.; Sachkova, M.Y.; Sykilinda, N.N.; Mesyanzhinov, V.V. Characterization of tail sheath protein of giant bacteriophage φKZ Pseudomonas aeruginosa. Virology 2009, 395, 312–317. [Google Scholar] [CrossRef]
- To, C.M.; Kellenberger, Y.; Eisenstark, A. Disassembly of T-even bacteriophage into structural parts and subunits. J. Mol. Biol. 1969, 46, 493–511. [Google Scholar] [CrossRef]
- Arisaka, F.; Engel, J.; Horst, K. Contraction and dissociation of the bacteriophage T4 tail sheath induced by heat and urea. Prog. Clin. Biol. Res. 1981, 64, 365–379. [Google Scholar]
- Arisaka, F.; Takeda, S.; Funane, K.; Nashijima, N.; Ishii, S. Structural studies of the contractile tail sheath protein of bacteriophage T4. Structural analyses of the tail sheath protein, gp18, by limited proteolysis, immunoblotting, and immunoelectron microscopy. Biochemistry 1990, 29, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Poglazov, B.F.; Efimov, A.V.; Marco, S.; Carrascosa, J.; Kuznetsova, T.A.; Aijrich, L.G.; Kurochkina, L.P.; Mesyanzhinov, V.V. Polymerization of bacteriophage T4 tail sheath protein mutants truncated at the C-termini. J. Struct. Biol. 1999, 127, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Efimov, V.P.; Kurochkina, L.P.; Mesyanzhinov, V.V. Engineering of bacteriophage T4 tail sheath protein. Biochemistry 2002, 67, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Truncaite, L.; Šimoliūnas, E.; Zajanckauskaite, A.; Kaliniene, L.; Mankeviciute, R.; Staniulis, J.; Klausa, V.; Meskys, R. Bacteriophage vB_EcoM_FV3: A new member of “rV5-like viruses”. Arch. Virol. 2012, 157, 2431–2435. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Hellman, U.; Wernstedt, C.; Gonez, J.; Heldin, C.H. Improvement of an ‘‘in-gel’’ digestion procedure for the micro preparation of internal protein fragments for amino acid sequencing. Anal. Biochem. 1995, 224, 451–455. [Google Scholar] [CrossRef]
- Ger, M.; Kaupinis, A.; Nemeikaite-Ceniene, A.; Sarlauskas, J.; Cicenas, J.; Cenas, N.; Valius, M. Quantitative proteomic analysis of anticancer drug RH1 resistance in liver carcinoma. Biochim. Biophys. Acta 2016, 1864, 219–232. [Google Scholar] [CrossRef]
- Povilonienė, S.; Časaitė, V.; Bukauskas, V.; Šetkus, A.; Staniulis, J.; Meškys, R. Functionalization of α-synuclein fibrils. Beilstein J. Nanotechnol. 2015, 6, 124–133. [Google Scholar] [CrossRef]
- Transeq. Available online: http://www.ebi.ac.uk/Tools/st/emboss_transeq (accessed on 10 January 2019).
- Clustal Omega. Available online: http://www.ebi.ac.uk/Tools/msa/clustalo (accessed on 10 January 2019).
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- PIR. Available online: http://pir.georgetown.edu/pirwww/search/comp_mw.shtml (accessed on 10 January 2019).
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- HHpred. Available online: https://toolkit.tuebingen.mpg.de/hhpred (accessed on 10 January 2019).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- I-TASSER. Available online: https://zhanglab.ccmb.med.umich.edu/I-TASSER (accessed on 10 January 2019).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- UCSF Chimera. Available online: http://www.rbvi.ucsf.edu/chimera (accessed on 10 January 2019).
- Aksyuk, A.A.; Kurochkina, L.P.; Fokine, A.; Forouhar, F.; Mesyanzhinov, V.V.; Tong, L.; Rossmann, M.G. Structural conservation of the myoviridae phage tail sheath protein fold. Structure 2011, 19, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Novacek, J.; Siborova, M.; Benesik, M.; Pantucek, R.; Doskar, J.; Plevka, P. Structure and genome release of Twort-like Myoviridae phage with a double-layered baseplate. Proc. Natl. Acad. Sci. USA 2016, 113, 9351–9356. [Google Scholar] [CrossRef] [PubMed]
- Sarris, P.F.; Ladoukakis, E.D.; Panopoulos, N.J.; Scoulica, E.V. A phage tail-derived element with wide distribution among both prokaryotic domains: A comparative genomic and phylogenetic study. Genome Biol. Evol. 2014, 6, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Kube, S.; Wendler, P. Structural comparison of contractile nanomachines. AIMS Biophys. 2015, 2, 88–115. [Google Scholar] [CrossRef]
- Kudryashev, M.; Wang, R.Y.; Brackmann, M.; Scherer, S.; Maier, T.; Baker, D.; DiMaio, F.; Stahlberg, H.; Egelman, E.H.; Basler, M. Structure of the type VI secretion system contractile sheath. Cell 2015, 160, 952–962. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; van Raaij, M.J.; Leiman, P.G. Contractile injection systems of bacteriophages and related systems. Mol. Microbiol. 2018, 108, 6–15. [Google Scholar] [CrossRef]
- Donelli, G.; Guglielmi, F.; Paoletti, L. Structure and physico-chemical properties of bacteriophage G. I. Arrangement of protein subunits and contraction process of tail sheath. J. Mol. Biol. 1972, 71, 113–125. [Google Scholar] [CrossRef]
- Cremers, A.F.M.; Schepman, A.M.H.; Visser, M.P.; Mellema, J.E. An analysis of the contracted sheath structure of bacteriophage Mu. Eur. J. Biochem. 1977, 80, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.L.; Eiserling, F.A. Bacteriophage SPO1 structure and morphogenesis. I. Tail structure and length regulation. J. Virol. 1983, 46, 239–249. [Google Scholar]
- Muller, M.; Engel, A.; Aebi, U. Structural and physicochemical analysis of the contractive MM phage tail and comparison with the bacteriophage T4 tail. J. Struct. Biol. 1994, 112, 11–31. [Google Scholar] [CrossRef]
- Fokine, A.; Battisti, A.J.; Bowman, V.D.; Efimov, A.V.; Kurochkina, L.P.; Chipman, P.R.; Mesyanzhinov, V.V.; Rossmann, M.G. Cryo-EM study of the Pseudomonas bacteriophage phiKZ. Structure 2007, 15, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Kurochkina, L.P.; Semenyuk, P.I.; Sykilinda, N.N.; Miroshnikov, K.A. The unique two-component tail sheath of giant Pseudomonas phage PaBG. Virology 2018, 515, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, T.A.; Efimov, A.V.; Aijrich, L.G.; Kireeva, I.Y.; Marusich, E.I.; Cappuccinelli, P.; Fiori, P.; Rappelli, P.; Kurochkina, L.P.; Poglazov, B.F.; et al. Properties of recombinant bacteriophage T4 tail sheath protein and its deletion fragments. Biokhimia 1998, 63, 702–709. [Google Scholar]
- Moody, M.F. Sheath of bacteriophage T4. III. Contraction mechanism deduced from partially contracted sheaths. J. Mol. Biol. 1973, 80, 613–635. [Google Scholar] [CrossRef]
- Peissker, T.; Deschaume, O.; Rand, D.R.; Boyen, H.G.; Conard, T.; Van Bael, M.J.; Bartic, C. Selective protein immobilization onto gold nanoparticles deposited under vacuum on a protein-repellent self-sssembled monolayer. Langmuir 2013, 29, 15328–15335. [Google Scholar] [CrossRef]
- Sharma, P.; Rathi, B.; Rodrigues, J.Y.; Gorobets, N. Self-assembled peptide nanoarchitectures: Applications and future aspects. Curr. Top. Med. Chem. 2015, 15, 1268–1289. [Google Scholar] [CrossRef]
- Glover, D.J.; Giger, L.; Kim, S.S.; Naik, R.R.; Clark, D.S. Geometrical assembly of ultrastable protein templates for nanomaterials. Nat. Commun. 2016, 7, 11771. [Google Scholar] [CrossRef]
- Knez, M.; Bittner, A.M.; Boes, F.; Wege, C.; Jeske, H.; Maiß, E.; Kern, K. Biotemplate synthesis of 3-nm nickel and cobalt nanowires. Nano Lett. 2003, 3, 1079–1082. [Google Scholar] [CrossRef]
- Bromley, K.M.; Patil, A.J.; Perriman, A.W.; Stubbs, G.; Mann, S. Preparation of high quality nanowires by tobacco mosaic virus templating of gold nanoparticles. J. Mater. Chem. 2008, 18, 4796–4801. [Google Scholar] [CrossRef]
- Warner, C.M.; Barker, N.; Lee, S.W.; Perkins, E.J. M13 bacteriophage production for large-scale applications. Bioprocess Biosyst. Eng. 2014, 37, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Lane, L.C. Propagation and purification of RNA plant viruses. Methods Enzymol. 1986, 118C, 687–696. [Google Scholar] [CrossRef]
- Shih, S.M.H.; Doran, P.M. In vitro propagation of plant virus using different forms of plant tissue culture and modes of culture operation. J. Biotechnol. 2009, 143, 198–206. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimoliūnas, E.; Truncaitė, L.; Rutkienė, R.; Povilonienė, S.; Goda, K.; Kaupinis, A.; Valius, M.; Meškys, R. The Robust Self-Assembling Tubular Nanostructures Formed by gp053 from Phage vB_EcoM_FV3. Viruses 2019, 11, 50. https://doi.org/10.3390/v11010050
Šimoliūnas E, Truncaitė L, Rutkienė R, Povilonienė S, Goda K, Kaupinis A, Valius M, Meškys R. The Robust Self-Assembling Tubular Nanostructures Formed by gp053 from Phage vB_EcoM_FV3. Viruses. 2019; 11(1):50. https://doi.org/10.3390/v11010050
Chicago/Turabian StyleŠimoliūnas, Eugenijus, Lidija Truncaitė, Rasa Rutkienė, Simona Povilonienė, Karolis Goda, Algirdas Kaupinis, Mindaugas Valius, and Rolandas Meškys. 2019. "The Robust Self-Assembling Tubular Nanostructures Formed by gp053 from Phage vB_EcoM_FV3" Viruses 11, no. 1: 50. https://doi.org/10.3390/v11010050
APA StyleŠimoliūnas, E., Truncaitė, L., Rutkienė, R., Povilonienė, S., Goda, K., Kaupinis, A., Valius, M., & Meškys, R. (2019). The Robust Self-Assembling Tubular Nanostructures Formed by gp053 from Phage vB_EcoM_FV3. Viruses, 11(1), 50. https://doi.org/10.3390/v11010050