The Oxysterol 7-Ketocholesterol Reduces Zika Virus Titers in Vero Cells and Human Neurons



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Virus Isolates
2.3. Reagents
2.4. Autophagy Compound Library Screen
2.5. Autophagy Detection
2.6. Cell Viability
2.7. Human Neuron Response to Autophagy Compounds
2.8. 7-Ketocholesterol Dose-Response
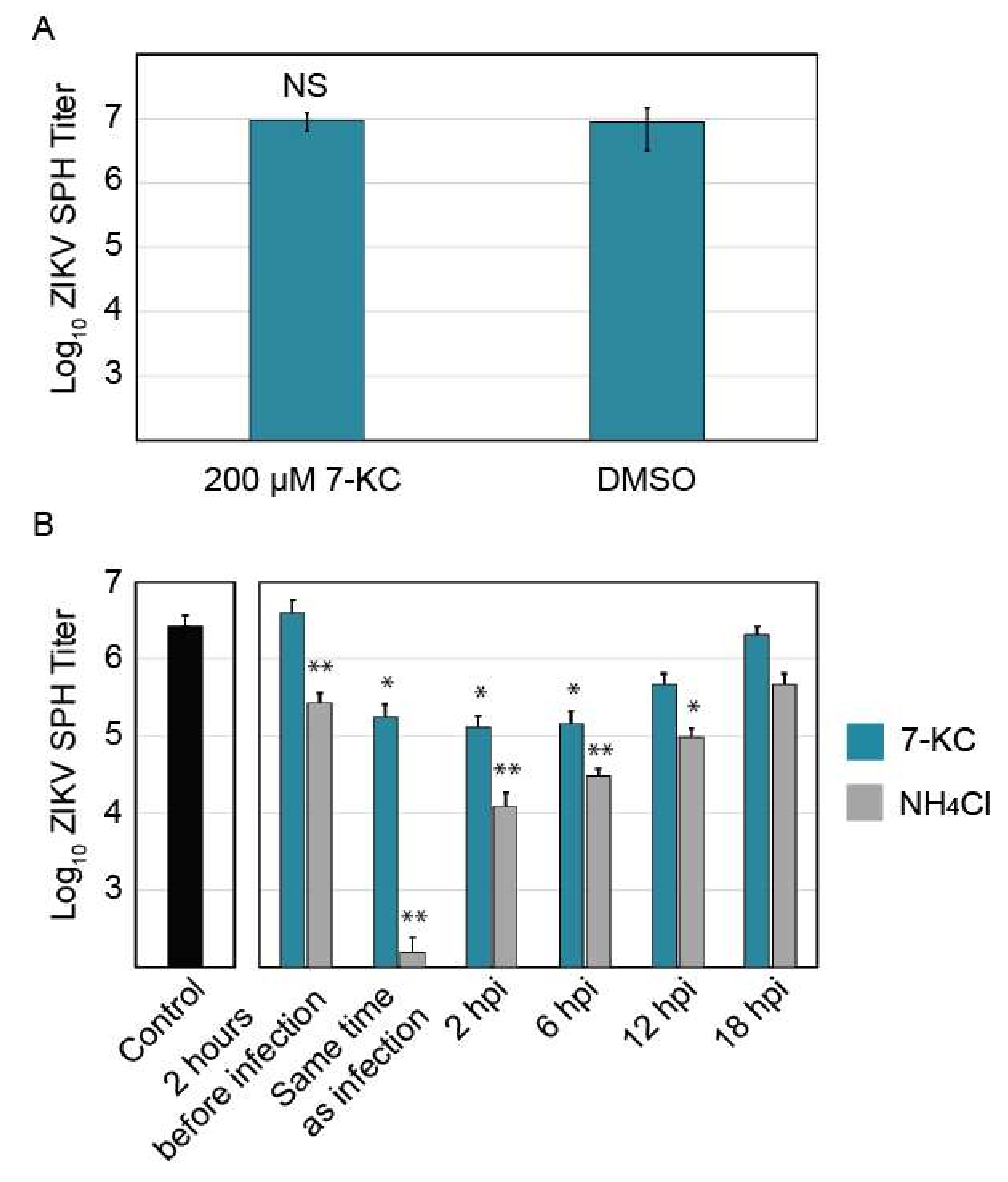
2.9. 7-Ketocholesterol Time-of-Addition
2.10. 7-Ketocholesterol and ZIKV Pre-Incubation
2.11. qRT-PCR
2.12. Chemical Structures
2.13. Statistics
3. Results
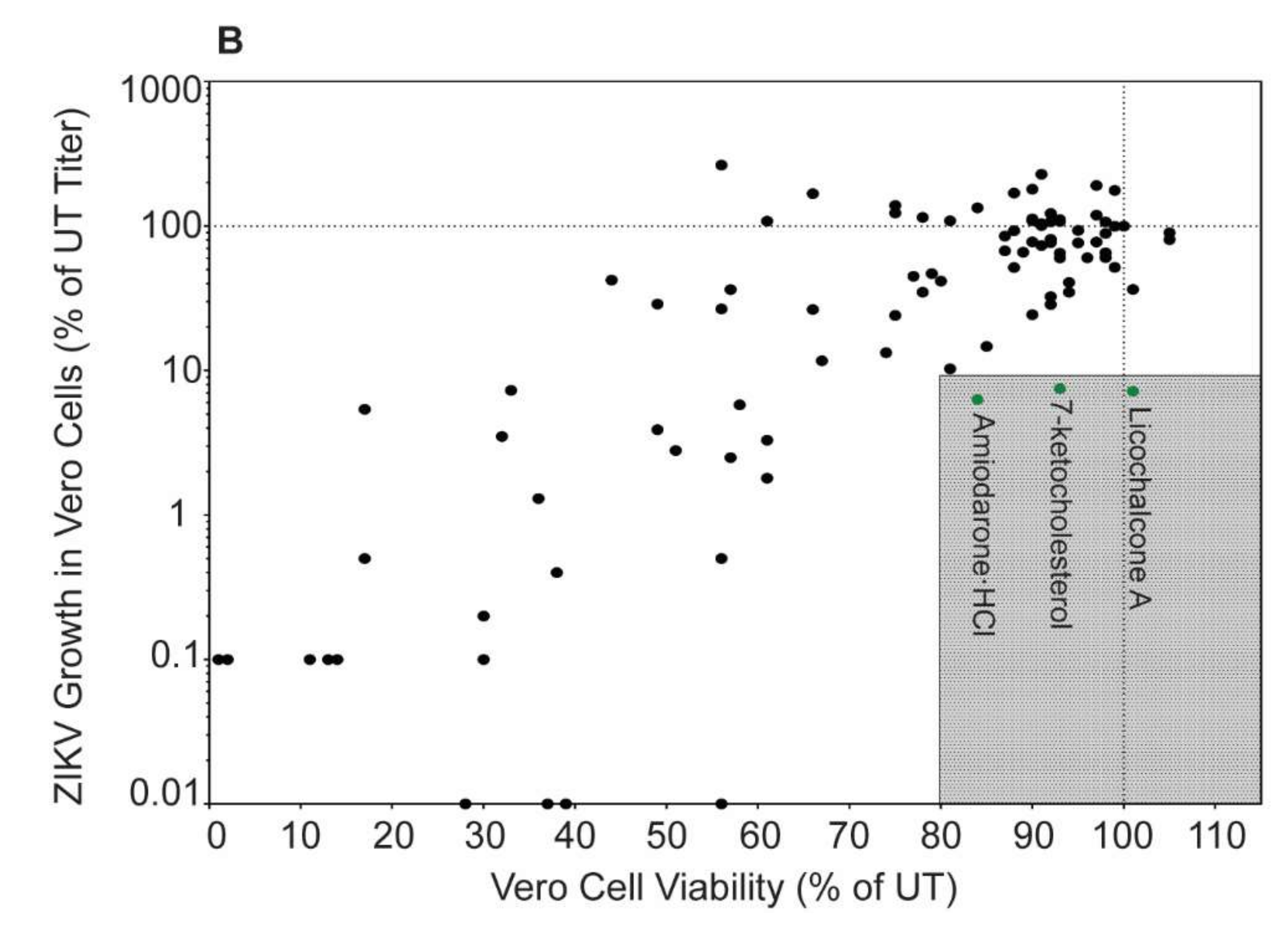
3.1. Autophagy Compound Screen in Vero Cells
3.2. Autophagy Compound Screen in Vero Cells Infected with ZIKV IbH
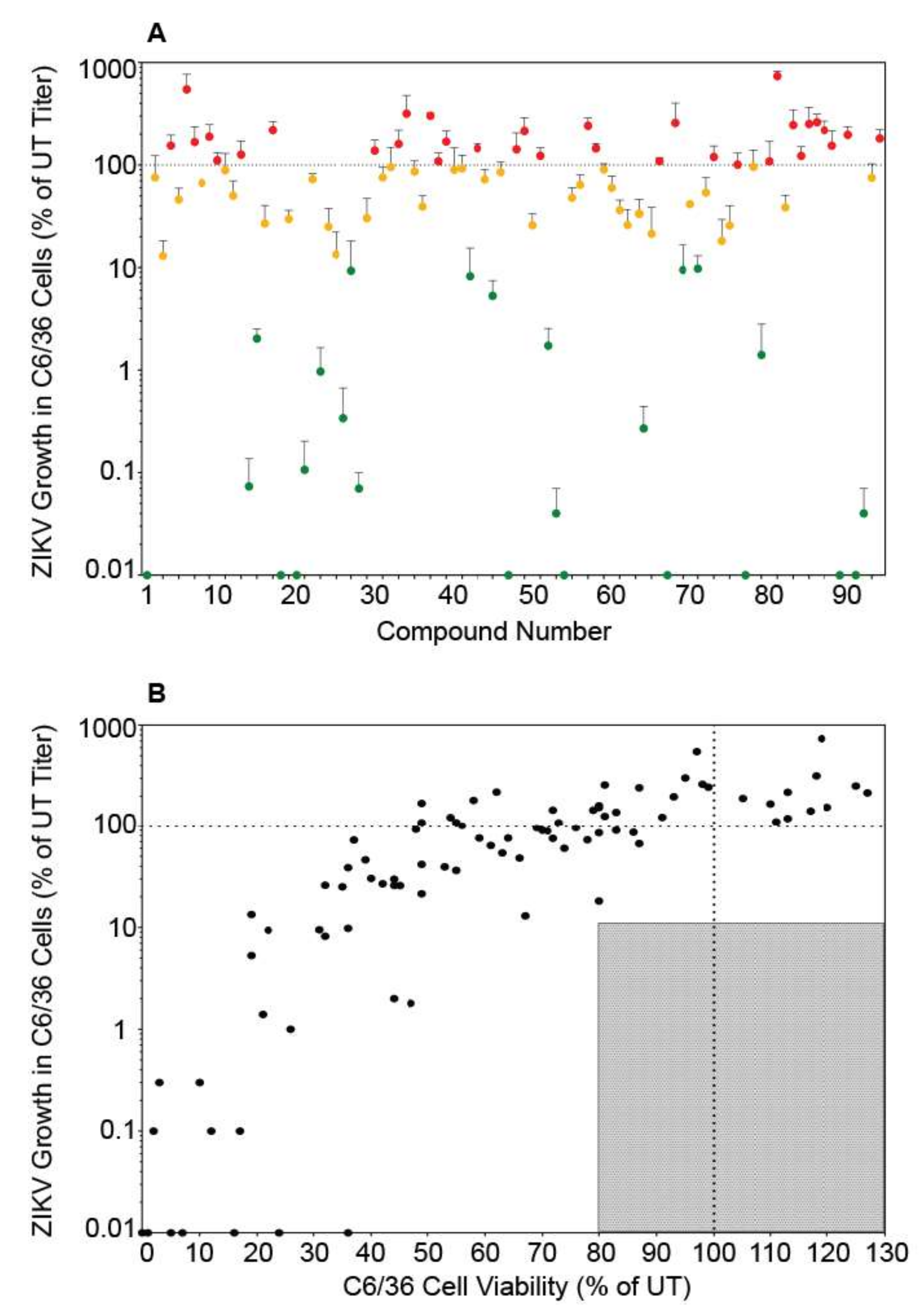
3.3. Autophagy Compound Screen in C6/36 Cells
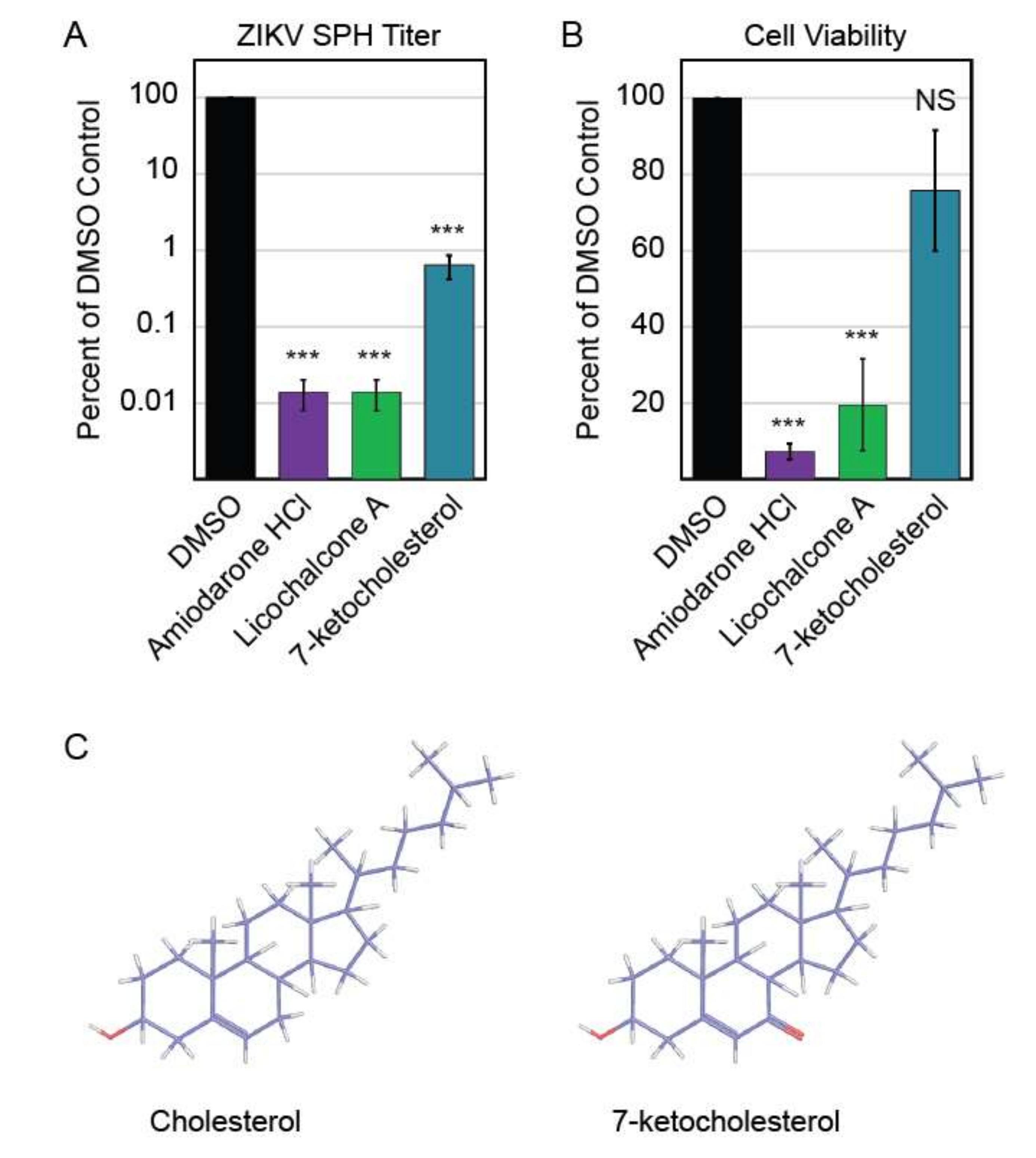
3.4. Autophagy Compound Screen in Human Neurons
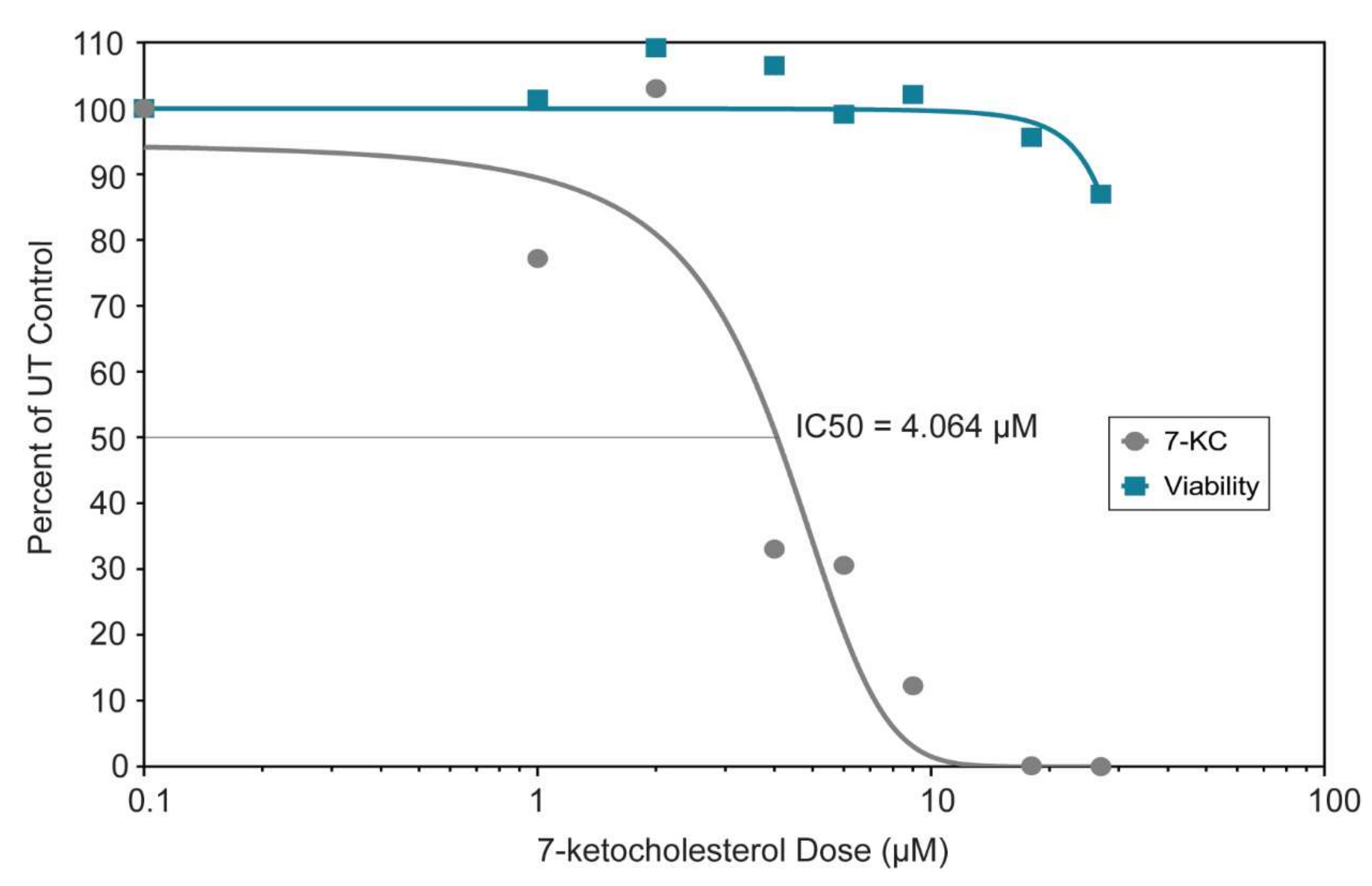
3.5. 7-Ketocholesterol Dose-Response
3.6. 7-Ketocholesterol Time-of-Addition
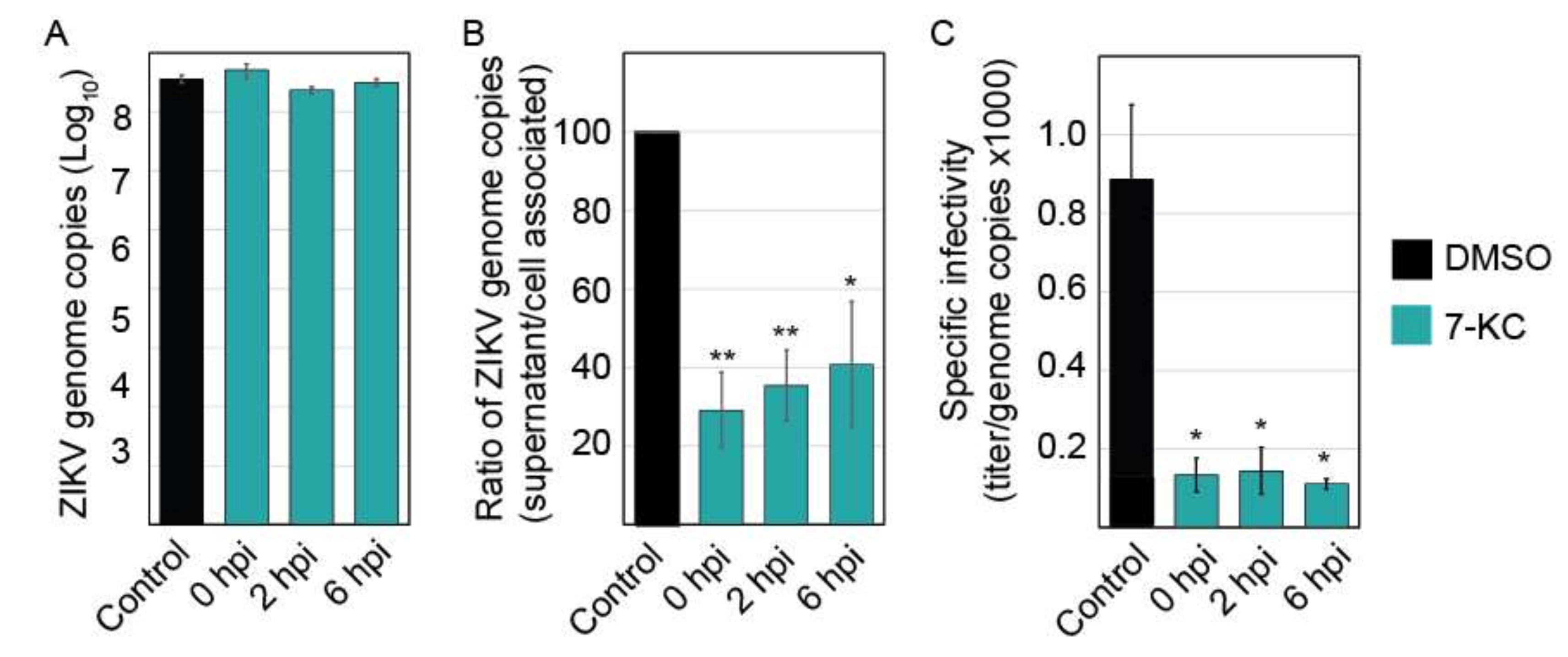
3.7. 7-Ketocholesterol qRT-PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broutet, N.; Krauer, F.; Riesen, M.; Khalakdina, A.; Almiron, M.; Aldighieri, S.; Espinal, M.; Low, N.; Dye, C. Zika Virus as a Cause of Neurologic Disorders. N. Engl. J. Med. 2016, 374, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Persaud, M.; Martinez-Lopez, A.; Buffone, C.; Porcelli, S.A.; Diaz-Griffero, F. Infection by Zika viruses requires the transmembrane protein AXL, endocytosis and low pH. Virology 2018, 518, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, E.D.; Peters, K.N.; Connor, J.H.; Bullitt, E. Zika virus induced cellular remodelling. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Knodler, L.A.; Celli, J. Eating the strangers within: Host control of intracellular bacteria via xenophagy. Cell. Microbiol. 2011, 13, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Stallings, C.L. Bacterial Pathogens versus Autophagy: Implications for Therapeutic Interventions. Trends Mol. Med. 2016, 22, 1060–1076. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. How positive-strand RNA viruses benefit from autophagosome maturation. J. Virol. 2013, 87, 9966–9972. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Li, Z.L.; Yuan, S. The Role of Secretory Autophagy in Zika Virus Transfer through the Placental Barrier. Front. Cell. Infect. Microbiol. 2016, 6, 206. [Google Scholar] [CrossRef]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.P.; Tsai, C.C.; Huang, W.C.; Wang, C.Y.; Chen, C.L.; Lin, Y.S.; Kai, J.I.; Hsieh, C.Y.; Cheng, Y.L.; Choi, P.C.; et al. Autophagy facilitates IFN-gamma-induced Jak2-STAT1 activation and cellular inflammation. J. Biol. Chem. 2010, 285, 28715–28722. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68.e3. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Mitalipova, M.; Noggle, S.; Tibbitts, D.; Venable, A.; Rao, R.; Stice, S.L. Long-term proliferation of human embryonic stem cell-derived neuroepithelial cells using defined adherent culture conditions. Stem Cells 2006, 24, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.A.; Russo, R.C.; Thurston, R.V. Trimmed Spearman-Karber Method for Estimating Median Lethal Concentrations in Toxicity Bioassays. Environ. Sci. Technol. 1977, 11, 714–719. [Google Scholar] [CrossRef]
- Spearman, C. The Method of ‘Right and Wrong Cases’ (‘Constant Stimuli’) without Gauss’s Formulae. Br. J. Psychol. 1908, 2, 227–242. [Google Scholar] [CrossRef]
- Chan, L.L.; Shen, D.; Wilkinson, A.R.; Patton, W.; Lai, N.; Chan, E.; Kuksin, D.; Lin, B.; Qiu, J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy 2012, 8, 1371–1382. [Google Scholar] [CrossRef]
- Goodfellow, F.T.; Willard, K.A.; Wu, X.; Scoville, S.; Stice, S.L.; Brindley, M.A. Strain-Dependent Consequences of Zika Virus Infection and Differential Impact on Neural Development. Viruses 2018, 10, 550. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Willard, K.A.; Demakovsky, L.; Tesla, B.; Goodfellow, F.T.; Stice, S.L.; Murdock, C.C.; Brindley, M.A. Zika Virus Exhibits Lineage-Specific Phenotypes in Cell Culture, in Aedes aegypti Mosquitoes, and in an Embryo Model. Viruses 2017, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Chen, Y.S.; Lin, C.C.; Chen, Y.J.; Lo, G.H.; Lee, P.H.; Kuo, P.L.; Dai, C.Y.; Huang, J.F.; Chung, W.L.; et al. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci. Rep. 2015, 5, 15807. [Google Scholar] [CrossRef] [PubMed]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic characterization of Zika virus strains: Geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 2012, 6, e1477. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Herrlinger, S.; Zhu, Y.N.; Yang, M.; Goodfellow, F.; Stice, S.L.; Qi, X.P.; Brindley, M.A.; Chen, J.F. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 2017, 144, 4114–4124. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; Loustalot, F.; Desmetz, C.; Foulongne, V.; Constant, O.; Fournier-Wirth, C.; Leon, F.; Moles, J.P.; Goubaud, A.; Lemaitre, J.M.; et al. Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine 2016, 12, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Moy, R.H.; Cherry, S. Antimicrobial autophagy: A conserved innate immune response in Drosophila. J. Innate Immun. 2013, 5, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-San Martin, C.; Liu, C.Y.; Kielian, M. Dealing with low pH: Entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009, 17, 514–521. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, Z.W.; Li, Z.L. Trehalose May Decrease the Transmission of Zika Virus to the Fetus by Activating Degradative Autophagy. Front. Cell. Infect. Microbiol. 2017, 7, 402. [Google Scholar] [CrossRef]
- Shinkyo, R.; Xu, L.; Tallman, K.A.; Cheng, Q.; Porter, N.A.; Guengerich, F.P. Conversion of 7-dehydrocholesterol to 7-ketocholesterol is catalyzed by human cytochrome P450 7A1 and occurs by direct oxidation without an epoxide intermediate. J. Biol. Chem. 2011, 286, 33021–33028. [Google Scholar] [CrossRef]
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis 1999, 142, 1–28. [Google Scholar] [CrossRef]
- Arca, M.; Natoli, S.; Micheletta, F.; Riggi, S.; Di Angelantonio, E.; Montali, A.; Antonini, T.M.; Antonini, R.; Diczfalusy, U.; Iuliano, L. Increased plasma levels of oxysterols, in vivo markers of oxidative stress, in patients with familial combined hyperlipidemia: Reduction during atorvastatin and fenofibrate therapy. Free Radic. Biol. Med. 2007, 42, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.R.; Larrayoz, I.M. Cholesterol oxidation in the retina: Implications of 7KCh formation in chronic inflammation and age-related macular degeneration. J. Lipid Res. 2010, 51, 2847–2862. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Chou, Y.C.; Mazumder, N.; Kao, F.J.; Nagy, L.D.; Guengerich, F.P.; Huang, C.; Lee, H.C.; Lai, P.S.; Ueng, Y.F. 7-Ketocholesterol induces P-glycoprotein through PI3K/mTOR signaling in hepatoma cells. Biochem. Pharmacol. 2013, 86, 548–560. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhu, H.; Zhang, W.; Okon, I.; Wang, Q.; Li, H.; Le, Y.Z.; Xie, Z. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am. J. Pathol. 2013, 183, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.A.; Brown, A.J. 7-Ketocholesterol. Int. J. Biochem. Cell Biol. 1999, 31, 369–375. [Google Scholar] [CrossRef]
- Huang, J.D.; Amaral, J.; Lee, J.W.; Rodriguez, I.R. 7-Ketocholesterol-induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS ONE 2014, 9, e100985. [Google Scholar] [CrossRef]
- Civra, A.; Cagno, V.; Donalisio, M.; Biasi, F.; Leonarduzzi, G.; Poli, G.; Lembo, D. Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol. Sci. Rep. 2014, 4, 7487. [Google Scholar] [CrossRef]
- Massey, J.B.; Pownall, H.J. The polar nature of 7-ketocholesterol determines its location within membrane domains and the kinetics of membrane microsolubilization by apolipoprotein A-I. Biochemistry 2005, 44, 10423–10433. [Google Scholar] [CrossRef]
- Magenau, A.; Benzing, C.; Proschogo, N.; Don, A.S.; Hejazi, L.; Karunakaran, D.; Jessup, W.; Gaus, K. Phagocytosis of IgG-coated polystyrene beads by macrophages induces and requires high membrane order. Traffic 2011, 12, 1730–1743. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.M.; Fairn, G.D. 7-Ketocholesterol impairs phagocytosis and efferocytosis via dysregulation of phosphatidylinositol 4,5-bisphosphate. Traffic 2018, 19, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, Y.Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.Y.; Zhang, N.N.; Watanabe, M.; Dong, H.L.; Liu, P.; et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Sager, G.; Gabaglio, S.; Sztul, E.; Belov, G.A. Role of Host Cell Secretory Machinery in Zika Virus Life Cycle. Viruses 2018, 10, 559. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # a | Name | Type b | ZIKV SPH Relative Titer in Vero Cells c | Vero Viability d | ZIKV SPH Relative Titer in C6/36 Cells c | C6/36 Viability d |
|---|---|---|---|---|---|---|
| 1 | Bafilomycin A1 | H | 0.0 ± 0 | 55.7 ± 9.5 | 0.0 ± 0 | 36 ± 6.1 |
| 2 | Rapamycin | D | 167.9 ± 44.4 | 66.0 ± 2.6 | 75.7 ± 47.7 | 59 ± 7.3 |
| 3 | Timosaponin A-III | D | 0.0 ± 0 | 38.7 ± 11.9 | 13.0 ± 5.2 | 67 ± 3.6 |
| 4 | 3-Methyladenine | H | 73.4 ± 4.9 | 90.7 ± 1.8 | 155.3 ± 41 | 120 ± 15.6 |
| 5 | PI-103 | D | 42.3 ± 20.6 | 43.7 ± 6.4 | 46.2 ± 13.3 | 39 ± 8.9 |
| 6 | LY294002 | H | 264.3 ± 182.9 | 56.0 ± 6 | 548.4 ± 221 | 97 ± 17.2 |
| 7 | Lithium Chloride | D | 85.4 ± 18.7 | 87.0 ± 1 | 167.9 ± 68.4 | 111 ± 10.7 |
| 8 | L-690,330 | D | 40.7 ± 10.6 | 93.7 ± 3.5 | 66.8 ± 2.6 | 87 ± 4.6 |
| 9 | Wortmannin | H | 46.9 ± 9 | 79.0 ± 3.1 | 190.1 ± 57.8 | 105 ± 5.5 |
| 10 | Sodium Valproate | D | 110.8 ± 80.4 | 92.7 ± 1.2 | 111.4 ± 20.7 | 111 ± 6.2 |
| 11 | Verapamil·HCl | D | 92.9 ± 20.8 | 87.7 ± 0.9 | 89.0 ± 40.8 | 71 ± 2.9 |
| 12 | SP600125 | H | 26.5 ± 15 | 66.0 ± 2.1 | 50.3 ± 19.8 | 132 ± 9 |
| 13 | Chloroquine | H | 109.0 ± 65 | 89.7 ± 4.4 | 126.5 ± 44.8 | 81 ± 17.1 |
| 14 | Loperamide HCl | D | 11.7 ± 3.3 | 66.7 ± 17.4 | 0.1 ± 0.1 | 12 ± 6.1 |
| 15 | Amiodarone HCl | D | 6.3 ± 3.6 | 84.0 ± 8.2 | 2.0 ± 0.5 | 44 ± 15.3 |
| 16 | Nimodipine | D | 32.5 ± 7.3 | 92.0 ± 1.5 | 26.9 ± 13.6 | 41 ± 8.4 |
| 17 | Nitrendipine | D | 66.0 ± 27.1 | 88.7 ± 1.9 | 219.7 ± 44.4 | 62 ± 10.5 |
| 18 | Niguldipine | D | 0.2 ± 0.1 | 30.0 ± 14.2 | 0.0 ± 0 | 5 ± 2.1 |
| 19 | Penitrem A | D | 34.9 ± 10.4 | 78.3 ± 6.4 | 29.8 ± 6.4 | 43 ± 8.1 |
| 20 | Ionomycin | D | 0.0 ± 0 | 27.7 ± 10.4 | 0.0 ± 0 | 1 ± 0.3 |
| 21 | Rotenone | D | 1.8 ± 0.9 | 61.3 ± 7.7 | 0.1 ± 0.1 | 17 ± 5.8 |
| 22 | TTFA | D | 24.4 ± 7.6 | 90.0 ± 3.5 | 72.8 ± 9.6 | 78 ± 19.9 |
| 23 | Fluspirilene | D | 2.5 ± 0.6 | 56.7 ± 16 | 1.0 ± 0.7 | 26 ± 10.3 |
| 24 | Hydroxychloroquine | H | 14.7 ± 7.3 | 85.3 ± 8.4 | 25.1 ± 12.6 | 35 ± 9.7 |
| 25 | Norclomipramine HCl | H | 3.9 ± 0.5 | 48.7 ± 22.9 | 13.4 ± 8.7 | 19 ± 8.9 |
| 26 | Trifluoperazine·2HCl | D | 2.8 ± 0.7 | 51.0 ± 22.1 | 0.3 ± 0.3 | 10 ± 3.8 |
| 27 | Sorafenib tosylate | D | 5.8 ± 1.8 | 58.3 ± 9.9 | 9.4 ± 8.8 | 23 ± 8.8 |
| 28 | Niclosamide | D | 0.1 ± 0 | 29.7 ± 3.8 | 0.1 ± 0 | 2 ± 0.3 |
| 29 | Rottlerin | D | 10.3 ± 5 | 80.7 ± 8.5 | 30.3 ± 16.9 | 40 ± 8.4 |
| 30 | Caffeine | D | 80.6 ± 20.9 | 104.7 ± 8.6 | 138.4 ± 37.7 | 83 ± 4.6 |
| 31 | Metformin·HCl | D | 122.3 ± 41.5 | 92.3 ± 4.2 | 75.8 ± 19.2 | 64 ± 10.3 |
| 32 | Clonidine·HCl | D | 65.4 ± 17.3 | 97.7 ± 2.6 | 95.9 ± 53.4 | 69 ± 5.4 |
| 33 | Rilmenidine | D | 191.2 ± 49.5 | 97.0 ± 2 | 161.0 ± 58.1 | 79 ± 8.3 |
| 34 | 2′,5′-Dideoxyadenosine | D | 93.3 ± 31.9 | 95.0 ± 1.5 | 317.4 ± 161.9 | 118 ± 15 |
| 35 | Suramin·6Na | D | 107.4 ± 44.8 | 92.0 ± 1.2 | 86.6 ± 23.2 | 86 ± 8.4 |
| 36 | (±)Bay K8644 | H | 34.9 ± 15.2 | 94.0 ± 3 | 39.4 ± 10.7 | 53 ± 11.1 |
| 37 | Forskolin | H | 139.0 ± 32.1 | 74.7 ± 3.7 | 302.5 ± 18.3 | 94 ± 12 |
| 38 | Pimozide | D | 77.9 ± 15.5 | 90.0 ± 5.7 | 109.0 ± 22 | 49 ± 3.6 |
| 39 | STF-62247 | D | 45.0 ± 21.4 | 77.3 ± 11.7 | 169.8 ± 45.9 | 49 ± 9.3 |
| 40 | Spermidine | D | 115.0 ± 44.9 | 78.0 ± 17 | 90.2 ± 57.4 | 70 ± 3 |
| 41 | FK-866 | D | 7.3 ± 6.5 | 32.7 ± 2.7 | 92.7 ± 32.4 | 48 ± 5.5 |
| 42 | Tamoxifen citrate | D | 3.3 ± 2.3 | 60.7 ± 18.4 | 8.2 ± 7.3 | 32 ± 11.9 |
| 43 | Minoxidil | D | 60.4 ± 27.2 | 96.3 ± 3.2 | 145.7 ± 16.6 | 72 ± 2.6 |
| 44 | Imiquimod | D | 228.5 ± 139.8 | 91.3 ± 1.2 | 72.5 ± 18.4 | 37 ± 8.1 |
| 45 | Imatinib mesylate | D | 91.2 ± 60 | 81.0 ± 10.7 | 5.3 ± 2.2 | 19 ± 5.4 |
| 46 | AG112 | D | 77.7 ± 38.2 | 96.7 ± 3.7 | 85.5 ± 21.3 | 80 ± 5.8 |
| 47 | SU11652 | D | 0.1 ± 0 | 1.0 ± 0 | 0.0 ± 0 | 1 ± 0 |
| 48 | Dibutyryl cAMP·Na | H | 60.6 ± 36.7 | 98.3 ± 4.4 | 142.6 ± 63.9 | 117 ± 10.7 |
| 49 | Rolipram | H | 103.8 ± 26.8 | 91.0 ± 3.2 | 215.4 ± 74.8 | 127 ± 12.1 |
| 50 | SB202190 | D | 13.3 ± 4.2 | 74.3 ± 5.2 | 25.9 ± 7.4 | 44 ± 7.9 |
| 51 | Brefeldin A | D | 5.4 ± 4.9 | 17.0 ± 2.1 | 123.5 ± 25 | 91 ± 8.4 |
| 52 | Tunicamycin | D | 0.5 ± 0.5 | 56.3 ± 8.7 | 1.8 ± 0.8 | 47 ± 7.6 |
| 53 | Thapsigargin | D | 0.4 ± 0.4 | 38.0 ± 10.5 | 0.0 ± 0 | 23 ± 3.9 |
| 54 | A23187 | D | 0.5 ± 0.5 | 17.0 ± 6.7 | 0.0 ± 0 | 1 ± 0 |
| 55 | Capsaicin | D | 76.9 ± 29.6 | 91.7 ± 2.4 | 48.1 ± 12 | 66 ± 7.2 |
| 56 | Dihydrocapsaicin | D | 28.7 ± 10.5 | 92.3 ± 3.8 | 64.1 ± 16 | 61 ± 2.2 |
| 57 | Glucosamine HCl | D | 51.8 ± 11.8 | 99.3 ± 1.9 | 242.1 ± 45.8 | 88 ± 5.2 |
| 58 | DTT | D | 60.2 ± 20.1 | 93.0 ± 1.5 | 145.7 ± 16.6 | 79 ± 4.4 |
| 59 | Deoxycholate·Na | D | 76.6 ± 16.6 | 95.3 ± 2.6 | 90.6 ± 12.5 | 83 ± 1 |
| 60 | 8-CPT-cAMP·Na | H | 107.8 ± 65.3 | 92.7 ± 0.7 | 60.1 ± 18.3 | 75 ± 3.7 |
| 61 | EHNA·HCl | H | 81.0 ± 7.6 | 92.3 ± 2 | 36.4 ± 9.1 | 55 ± 4.2 |
| 62 | ABC294640·HCl | D | 51.7 ± 29.2 | 87.7 ± 6.8 | 26.0 ± 10.8 | 32 ± 3.5 |
| 63 | Licochalcone A | D | 7.2 ± 2.9 | 100.7 ± 9.4 | 51.8 ± 28.5 | 43 ± 2 |
| 64 | Curcumin | D | 24.1 ± 11.3 | 75.3 ± 11.3 | 0.3 ± 0.2 | 3 ± 0.7 |
| 65 | Plumbagin | D | 0.1 ± 0 | 2.0 ± 0 | 21.4 ± 17.5 | 49 ± 1.5 |
| 66 | 6-Gingerol | D | 99.9 ± 63.9 | 100.0 ± 2.5 | 109.3 ± 9.3 | 55 ± 11 |
| 67 | Akt Inhibitor X·HCl | D | 3.5 ± 3.3 | 32.3 ± 14.2 | 0.0 ± 0 | 7 ± 1.9 |
| 68 | PMSF | D | 89.0 ± 7 | 98.3 ± 2 | 258.0 ± 145.2 | 81 ± 6.4 |
| 69 | MG132 | D | 0.1 ± 0.1 | 10.7 ± 2.8 | 9.5 ± 7.1 | 31 ± 5.7 |
| 70 | ALLN | D | 123.3 ± 27.3 | 75.0 ± 17.1 | 41.6 ± 0.6 | 49 ± 6.6 |
| 71 | 7-Ketocholesterol | D | 7.5 ± 1.3 | 93.0 ± 4 | 9.8 ± 3.3 | 36 ± 5.1 |
| 72 | SB-216763 | H | 41.6 ± 30.6 | 80.0 ± 9 | 54.0 ± 21.7 | 63 ± 3.6 |
| 73 | Tolazamide | H | 176.8 ± 50.9 | 99.0 ± 1.5 | 120.1 ± 33.7 | 113 ± 7.3 |
| 74 | 17-AAG | D | 36.4 ± 20.8 | 56. 7 ± 11.8 | 18.2 ± 11.2 | 79 ± 16.4 |
| 75 | Geldanamycin | D | 108.4 ± 39.4 | 61.3 ± 5.7 | 25.8 ± 14.5 | 45 ± 5.8 |
| 76 | C1 | D | 119.3 ± 34.4 | 97.3 ± 2.4 | 101.2 ± 29.5 | 55 ± 1.7 |
| 77 | Z36 | D | 0.1 ± 0.1 | 0.7 ± 0.3 | 0.0 ± 0 | 0 ± 0 |
| 78 | Rockout | D | 169.6 ± 69.3 | 87.7 ± 3.8 | 96.2 ± 44.2 | 76 ± 5.2 |
| 79 | Go6850 | D | 1.3 ± 0.9 | 36.0 ± 9 | 1.4 ± 1.4 | 21 ± 0 |
| 80 | 2-Deoxyglucose | D | 99.7 ± 15.4 | 98.7 ± 0.9 | 108.8 ± 61.2 | 73 ± 1 |
| 81 | Etoposide | D | 134.0 ± 34.3 | 84.0 ± 3.6 | 736.8 ± 89.3 | 119 ± 12.5 |
| 82 | SMER28 | D | 65.0 ± 5.6 | 92.7 ± 3.5 | 38.7 ± 12.2 | 36 ± 5.3 |
| 83 | Trehalose | D | 107.1 ± 16.9 | 97.7 ± 1.5 | 245.7 ± 101.3 | 99 ± 2.9 |
| 84 | Quinine HCl·2H2O | H | 180.2 ± 70.4 | 89.7 ± 2.7 | 123.0 ± 28 | 54 ± 4.6 |
| 85 | AICAR | H | 112.3 ± 32.3 | 90.0 ± 11.1 | 252.0 ± 115 | 125 ± 16.4 |
| 86 | C2-dihydroceramide | D | 36.5 ± 3 | 101.3 ± 3.7 | 262.1 ± 51.7 | 97 ± 5.8 |
| 87 | Temozolomide | D | 89.9 ± 17.5 | 104.7 ± 1.9 | 218.7 ± 49.8 | 113 ± 9.2 |
| 88 | Resveratrol | D | 67.5 ± 13.2 | 87.3 ± 2.3 | 154.6 ± 62 | 81 ± 7.5 |
| 89 | Staurosporine | D | 0.1 ± 0 | 14.0 ± 1.5 | 0.0 ± 0 | 16 ± 5.3 |
| 90 | PD-98059 | H | 101.0 ± 23.3 | 90.7 ± 1.3 | 197.3 ± 40 | 93 ± 8.4 |
| 91 | Anisomycin | H | 0.0 ± 0 | 12.7 ± 4.3 | 0.0 ± 0 | 7 ± 0.9 |
| 92 | Cycloheximide | H | 0.0 ± 0 | 37.3 ± 6.2 | 0.0 ± 0 | 24 ± 3.5 |
| 93 | Pifithrin-µ | H | 38.5 ± 12.4 | 49.0 ± 15 | 75.3 ± 27.5 | 72 ± 3.2 |
| 94 | Nocodazole | H | 28.4 ± 23.9 | 56.0 ± 9.6 | 182.0 ± 39.5 | 58 ± 2.9 |
| Compound | Autophagy Activity Factor (AAF) |
|---|---|
| DMSO | −0.85 |
| Rapamycin | 7.74 |
| Amiodarone HCl | 60.40 |
| Licochalcone A | 12.33 |
| 7-Ketocholesterol | 27.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willard, K.A.; Elling, C.L.; Stice, S.L.; Brindley, M.A. The Oxysterol 7-Ketocholesterol Reduces Zika Virus Titers in Vero Cells and Human Neurons. Viruses 2019, 11, 20. https://doi.org/10.3390/v11010020
Willard KA, Elling CL, Stice SL, Brindley MA. The Oxysterol 7-Ketocholesterol Reduces Zika Virus Titers in Vero Cells and Human Neurons. Viruses. 2019; 11(1):20. https://doi.org/10.3390/v11010020
Chicago/Turabian StyleWillard, Katherine A., Christina L. Elling, Steven L. Stice, and Melinda A. Brindley. 2019. "The Oxysterol 7-Ketocholesterol Reduces Zika Virus Titers in Vero Cells and Human Neurons" Viruses 11, no. 1: 20. https://doi.org/10.3390/v11010020
APA StyleWillard, K. A., Elling, C. L., Stice, S. L., & Brindley, M. A. (2019). The Oxysterol 7-Ketocholesterol Reduces Zika Virus Titers in Vero Cells and Human Neurons. Viruses, 11(1), 20. https://doi.org/10.3390/v11010020