Viperin Inhibits Enterovirus A71 Replication by Interacting with Viral 2C Protein


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus, Cell Culture, Transfection, and Infection
2.2. Antibodies and Sirna
2.3. Plasmids
2.4. Animal Models
2.5. Immunohistochemistry
2.6. Co-IP and Immunoblotting Assays
2.7. RNA Extraction and Reverse Transcription–Quantitative PCR (RT-qPCR) Analysis
2.8. Immunofluorescence and Confocal Microscopy
2.9. Statistical Analysis
3. Results
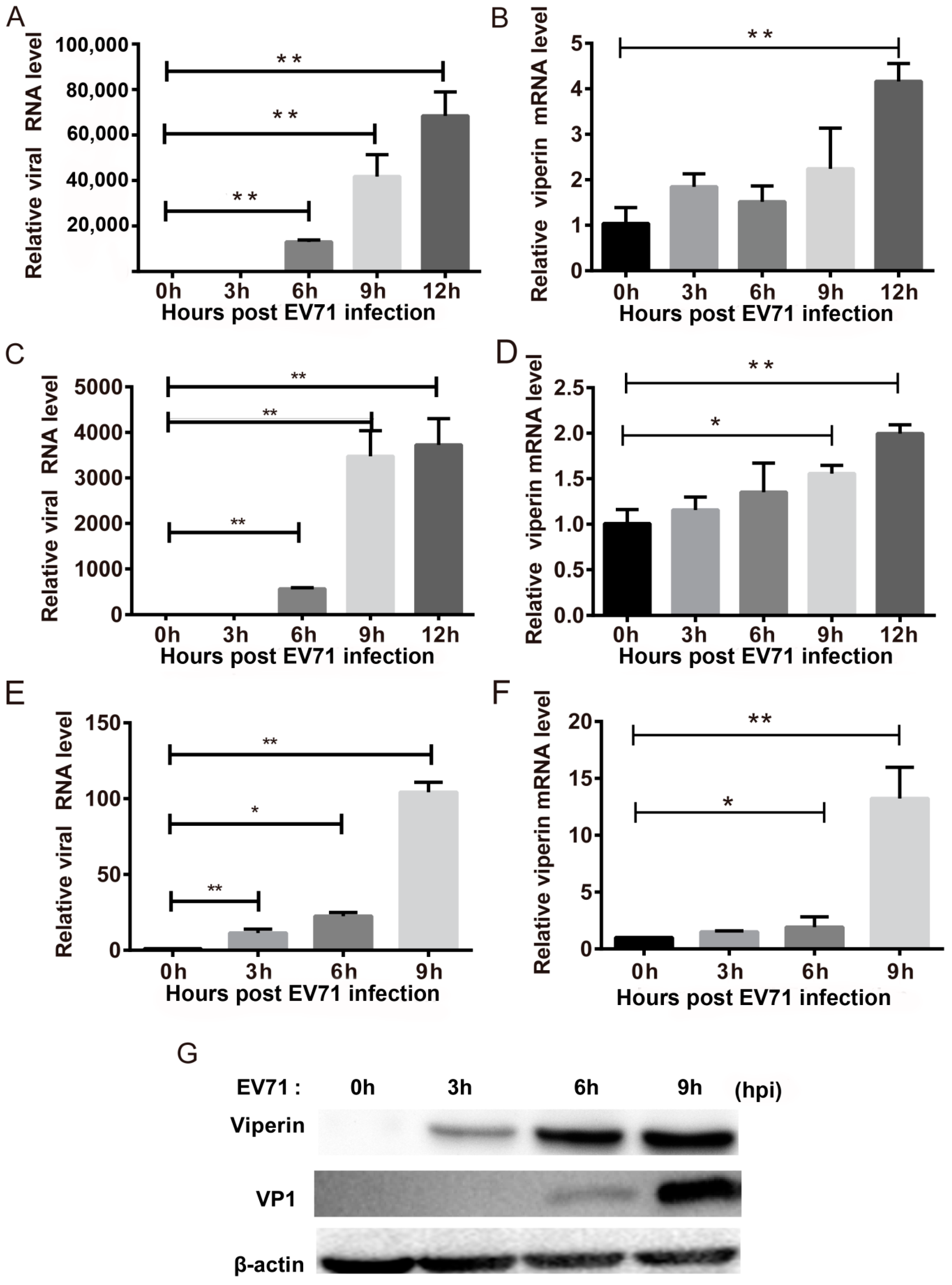
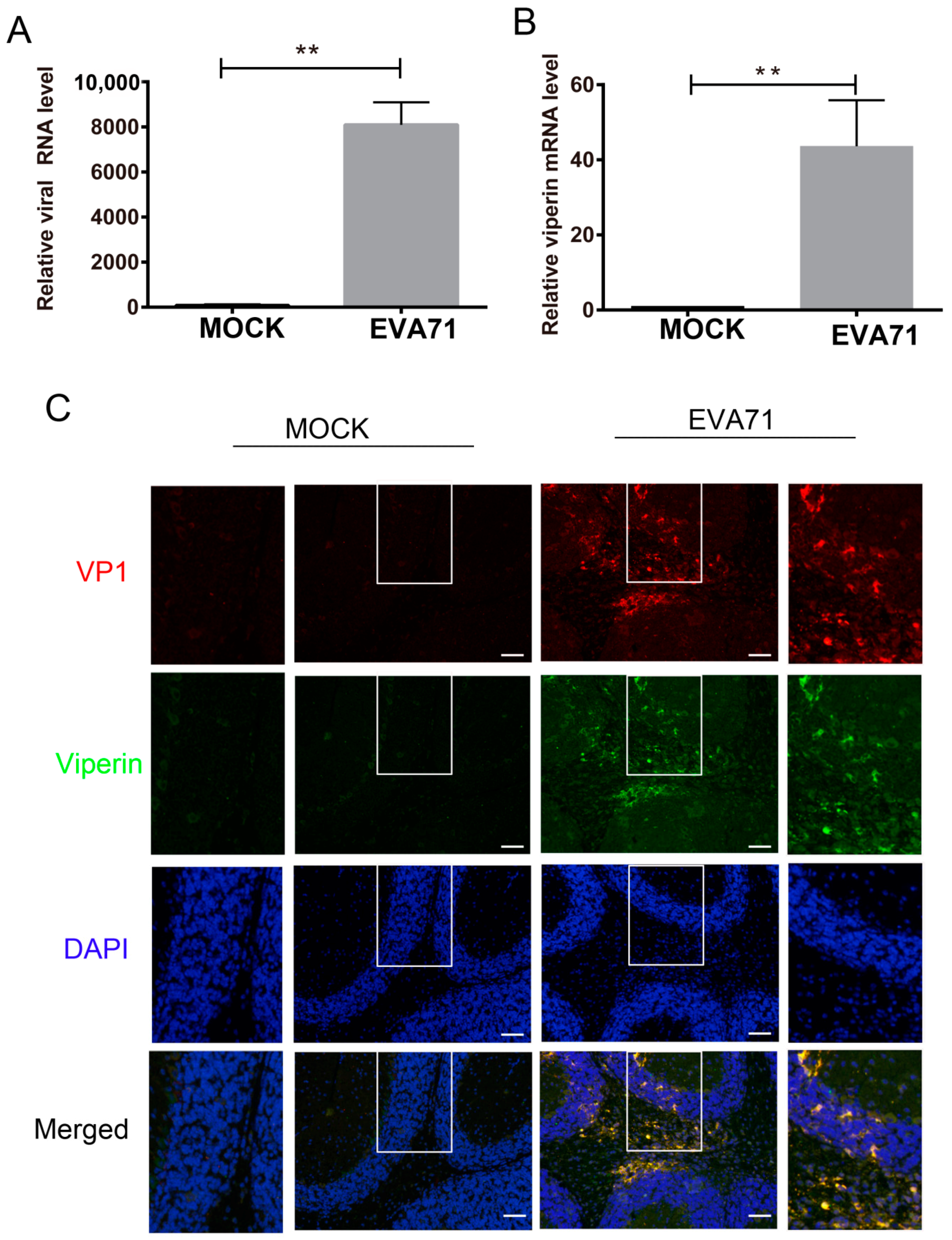
3.1. EVA71 Induced Viperin Expression In Vitro and In Vivo
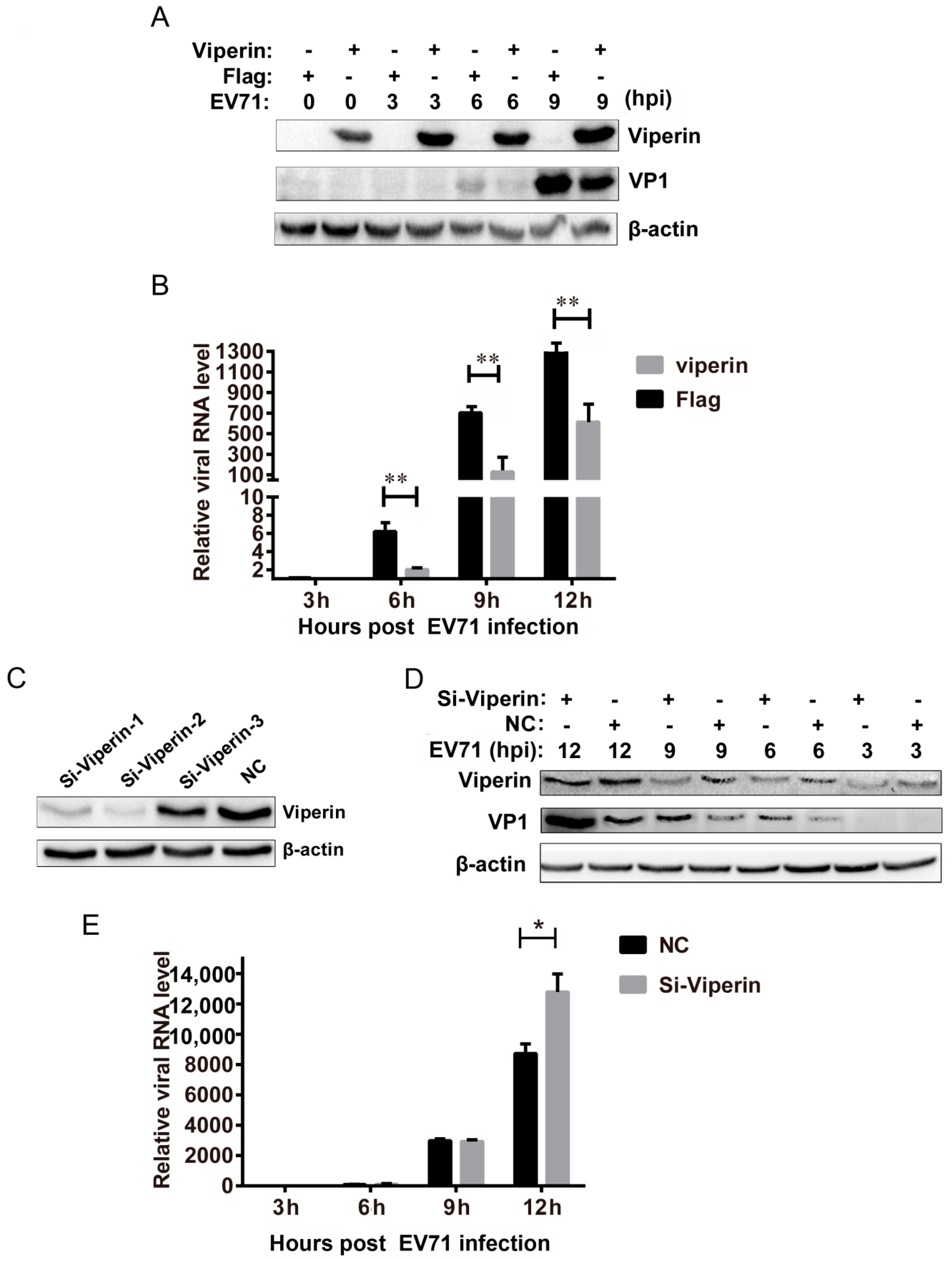
3.2. Viperin Inhibited EVA71 Replication
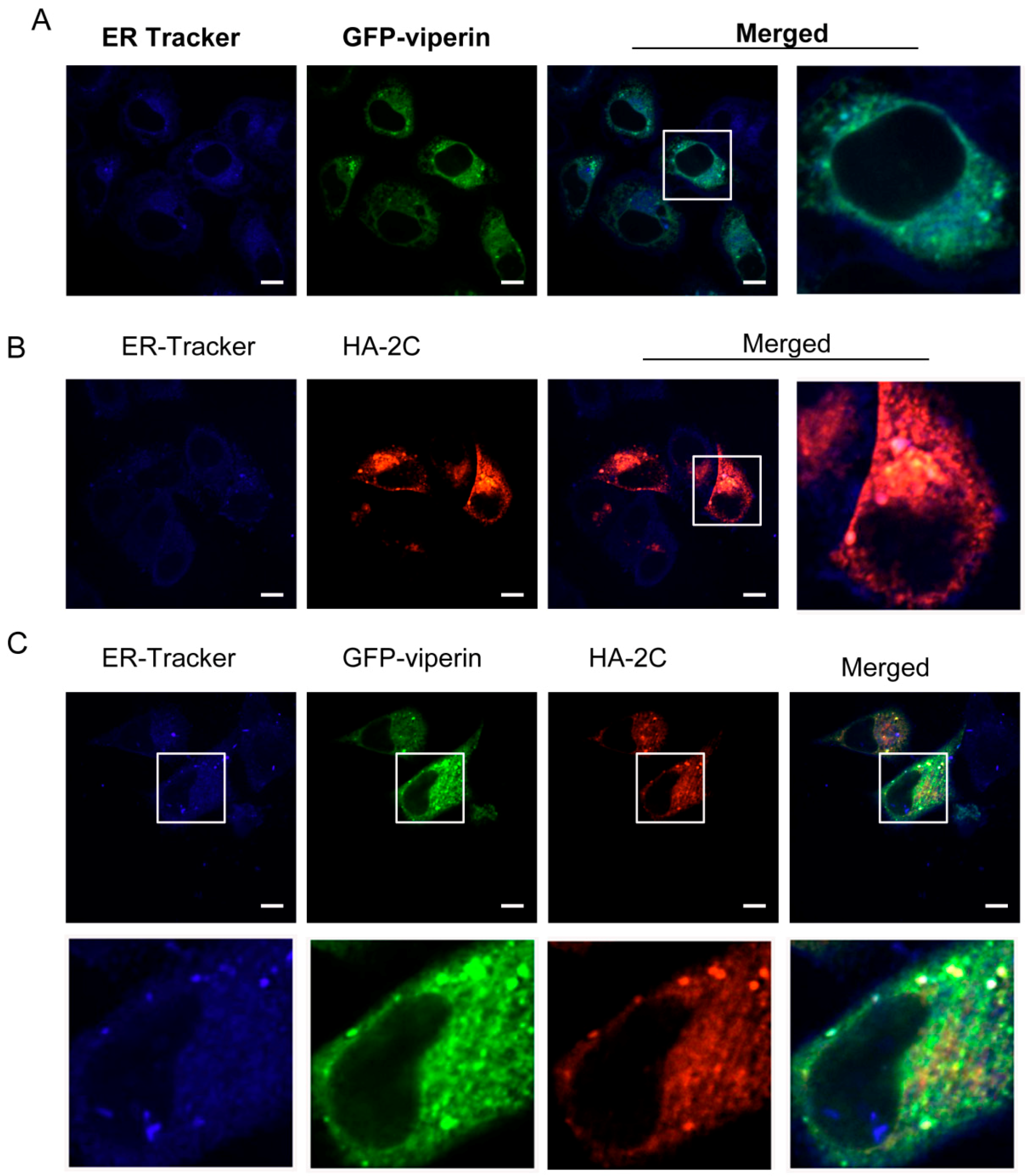
3.3. Viperin Was Colocalized with EVA71 2C in the ER
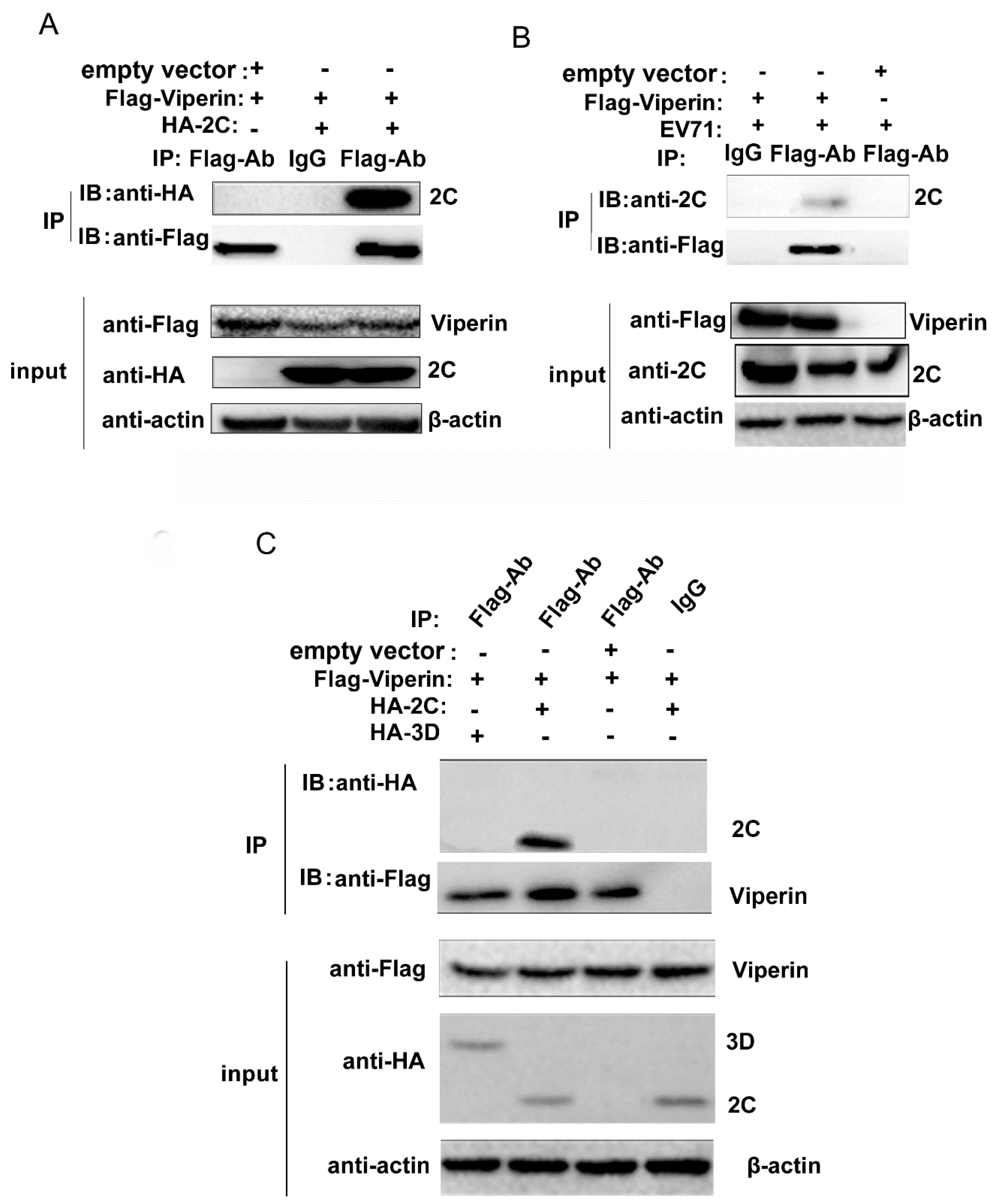
3.4. EVA71 2C Interacted with Viperin
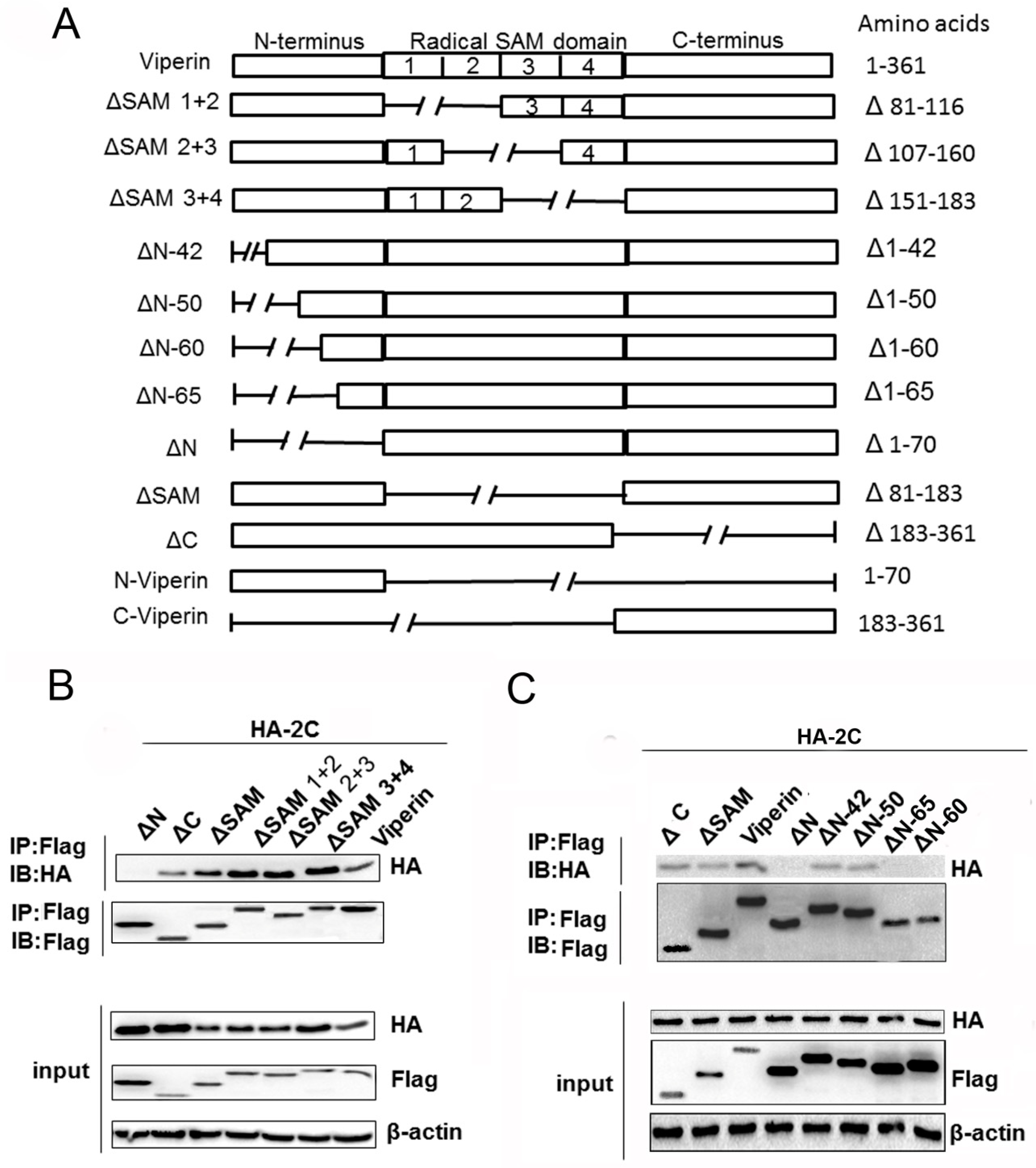
3.5. The N-Terminal Domain Was Responsible for Viperin Binding to 2C
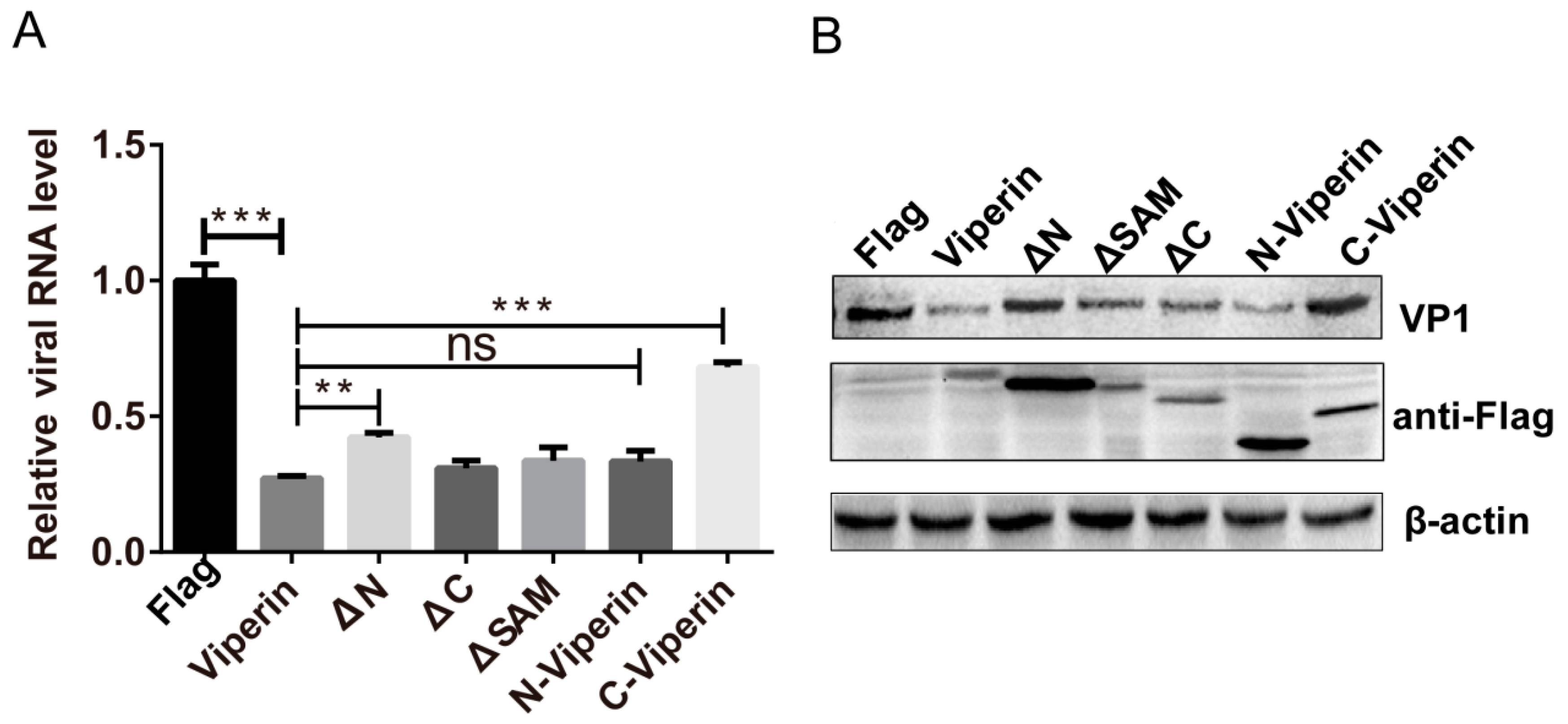
3.6. The N-Terminal Domain Was Responsible for Viperin Inhibition of EVA71 Replication
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uematsu, S.; Akira, S. Innate immune recognition of viral infection. Uirusu 2006, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The role of viperin in the innate antiviral response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Pallansch, M.; Roos, R. Fields Virology; Lippicott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 1, pp. 723–775. [Google Scholar]
- Xia, H.; Wang, P.; Wang, G.-C.; Yang, J.; Sun, X.; Wu, W.; Qiu, Y.; Shu, T.; Zhao, X.; Yin, L. Human enterovirus nonstructural protein 2CATPase functions as both an RNA helicase and ATP-independent RNA chaperone. PLoS Pathog. 2015, 11, e1005067. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, C.; Mueller, S.; Paul, A.V.; Wimmer, E.; Jiang, P. Direct interaction between two viral proteins, the nonstructural protein 2CATPase and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog. 2010, 6, e1001066. [Google Scholar] [CrossRef]
- Tang, W.-F.; Yang, S.-Y.; Wu, B.-W.; Jheng, J.-R.; Chen, Y.-L.; Shih, C.-H.; Lin, K.-H.; Lai, H.-C.; Tang, P.; Horng, J.-T. Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J. Biol. Chem. 2007, 282, 5888–5898. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, H.; Zhang, Z.; Meng, J.; Mao, D.; Bai, B.; Lu, B.; Mao, P.; Hu, Q.; Wang, H. Enterovirus 71 2C protein inhibits TNF-α-mediated activation of NF-κB by suppressing IκB kinase β phosphorylation. J. Immunol. 2011, 187, 2202–2212. [Google Scholar] [CrossRef]
- Fitzgerald, K.A. The interferon inducible gene: Viperin. J. Interferon Cytokine Res. 2011, 31, 131–135. [Google Scholar] [CrossRef]
- Wang, X.Y.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Nasr, N.; Maddocks, S.; Turville, S.G.; Harman, A.N.; Woolger, N.; Helbig, K.J.; Wilkinson, J.; Bye, C.R.; Wright, T.K.; Rambukwelle, D.; et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 2012, 120, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Carr, J.M.; Calvert, J.K.; Wati, S.; Clarke, J.N.; Eyre, N.S.; Narayana, S.K.; Fiches, G.N.; McCartney, E.M.; Beard, M.R. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLoS Negl. Trop. Dis. 2013, 7, e2178. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, X.; Pan, T.; Song, W.; Wang, Y.; Zhang, F.; Yuan, Z. Viperin inhibits hepatitis C virus replication by interfering with binding of NS5A to host protein hVAP-33. J. Gen. Virol. 2012, 93, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.C.; Cresswell, P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2001, 98, 15125–15130. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Yaneva, R.; Hinson, E.R.; Cresswell, P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 2011, 332, 1093–1097. [Google Scholar] [CrossRef]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef]
- Zhang, Y.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 2007, 81, 11246–11255. [Google Scholar] [CrossRef]
- Rivieccio, M.A.; Suh, H.-S.; Zhao, Y.; Zhao, M.-L.; Chin, K.C.; Lee, S.C.; Brosnan, C.F. TLR3 ligation activates an antiviral response in human fetal astrocytes: A role for viperin/cig5. J. Immunol. 2006, 177, 4735–4741. [Google Scholar] [CrossRef]
- Teng, T.S.; Foo, S.S.; Simamarta, D.; Lum, F.M.; Teo, T.H.; Lulla, A.; Yeo, N.K.W.; Koh, E.G.L.; Chow, A.; Leo, Y.S. Viperin restricts chikungunya virus replication and pathology. J. Clin. Investig. 2012, 122, 4447–4460. [Google Scholar] [CrossRef]
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Overby, A.K. Viperin Restricts Zika Virus and Tick-Borne Encephalitis Virus Replication by Targeting NS3 for Proteasomal Degradation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Vonderstein, K.; Nilsson, E.; Hubel, P.; Nygard Skalman, L.; Upadhyay, A.; Pasto, J.; Pichlmair, A.; Lundmark, R.; Overby, A.K. Viperin targets flavivirus virulence by inducing assembly of non-infectious capsid particles. J. Virol. 2018, 92, e01751-17. [Google Scholar] [CrossRef]
- Gizzi, A.S.; Grove, T.L.; Arnold, J.J.; Jose, J.; Jangra, R.K.; Garforth, S.J.; Du, Q.; Cahill, S.M.; Dulyaninova, N.G.; Love, J.D.; et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558, 610–614. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, Z.; Shu, B.; Meng, J.; Zhang, Y.; Zheng, C.; Ke, X.; Gong, P.; Hu, Q.; Wang, H. SUMO Modification Stabilizes Enterovirus 71 Polymerase 3D To Facilitate Viral Replication. J. Virol. 2016, 90, 10472–10485. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, S.; Zhu, L.; Huang, Z.; Chen, L.; Xu, Y.; Yin, H.; Peng, T.; Wang, Y. Clinically isolated enterovirus A71 subgenogroup C4 strain with lethal pathogenicity in 14-day-old mice and the application as an EV-A71 mouse infection model. Antivir. Res. 2017, 137, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hinson, E.R.; Cresswell, P. The N-terminal amphipathic alpha-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. J. Biol. Chem. 2009, 284, 4705. [Google Scholar] [CrossRef] [PubMed]
- Teterina, N.L.; Gorbalenya, A.E.; Egger, D.; Bienz, K.; Ehrenfeld, E. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J. Virol. 1997, 71, 8962–8972. [Google Scholar] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Liu, M.L.; Lee, Y.P.; Wang, Y.F.; Lei, H.Y.; Liu, C.C.; Wang, S.M.; Su, I.J.; Wang, J.R.; Yeh, T.M.; Chen, S.H.; et al. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 2005, 86, 3263–3629. [Google Scholar] [CrossRef]
- Yi, L.; He, Y.; Chen, Y.; Kung, H.F.; He, M.L. Potent inhibition of human enterovirus 71 replication by type I interferon subtypes. Antivir. Ther. 2011, 16, 51–58. [Google Scholar] [CrossRef]
- Seo, J.-Y.; Yaneva, R.; Cresswell, P. Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 2011, 10, 534–539. [Google Scholar] [CrossRef]
- Yogarajah, T.; Ong, K.C.; Perera, D.; Wong, K.T. RSAD2 and AIM2 Modulate Coxsackievirus A16 and Enterovirus A71 Replication in Neuronal Cells in Different Ways That May Be Associated with Their 5′ Nontranslated Regions. J. Virol. 2018, 92, e01914–e01917. [Google Scholar] [CrossRef] [PubMed]
- Mattijssen, S.; Pruijn, G.J. Viperin, a key player in the antiviral response. Microbes Infect. 2012, 14, 419–426. [Google Scholar] [CrossRef]
- Stirnweiss, A.; Ksienzyk, A.; Klages, K.; Rand, U.; Grashoff, M.; Hauser, H.; Kröger, A. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J. Immunol. 2010, 184, 5179–5185. [Google Scholar] [CrossRef]
- DeFilippis, V.R.; Robinson, B.; Keck, T.M.; Hansen, S.G.; Nelson, J.A.; Früh, K.J. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J. Virol. 2006, 80, 1032–1037. [Google Scholar] [CrossRef]
- Grandvaux, N.; Servant, M.J.; Sen, G.C.; Balachandran, S.; Barber, G.N.; Lin, R.; Hiscott, J. Transcriptional profiling of interferon regulatory factor 3 target genes: Direct involvement in the regulation of interferon-stimulated genes. J. Virol. 2002, 76, 5532–5539. [Google Scholar] [CrossRef]
- Tan, K.S.; Olfat, F.; Phoon, M.C.; Hsu, J.P.; Howe, J.L.; Seet, J.E.; Chin, K.C.; Chow, V.T. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol. 2012, 93, 1269–1277. [Google Scholar] [CrossRef]
- Upadhyay, A.S.; Vonderstein, K.; Pichlmair, A.; Stehling, O.; Bennett, K.L.; Dobler, G.; Guo, J.T.; Supertifurga, G.; Lill, R.; AK, Ö. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cell. Microbiol. 2014, 16, 834–848. [Google Scholar] [CrossRef]
- Fenwick, M.K.; Li, Y.; Cresswell, P.; Modis, Y.; Ealick, S.E. Structural studies of viperin, an antiviral radical SAM enzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 6806–6811. [Google Scholar] [CrossRef] [PubMed]
- Makins, C.; Ghosh, S.; Román-Meléndez, G.D.; Malec, P.A.; Kennedy, R.T.; Marsh, E.N. Does Viperin Function as a Radical S-Adenosyl-l-methionine-dependent Enzyme in Regulating Farnesylpyrophosphate Synthase Expression and Activity? J. Biol. Chem. 2016, 291, 26806. [Google Scholar] [CrossRef]
- Duschene, K.S.; Broderick, J.B. The antiviral protein viperin is a radical SAM enzyme. FEBS Lett. 2010, 584, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Carlton-Smith, C.; Elliott, R.M. Viperin, MTAP44, and protein kinase R contribute to the interferon-induced inhibition of Bunyamwera Orthobunyavirus replication. J. Virol. 2012, 86, 11548–11557. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nature reviews. Microbiology 2008, 6, 363–374. [Google Scholar] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Bárcena, M. The Transformation of Enterovirus Replication Structures: A Three-Dimensional Study of Single- and Double-Membrane Compartments. mBio 2011, 2, e00166-11. [Google Scholar] [CrossRef] [PubMed]
- Proud, D.; Turner, R.B.; Winther, B.; Wiehler, S.; Tiesman, J.P.; Reichling, T.D.; Juhlin, K.D.; Fulmer, A.W.; Ho, B.Y.; Walanski, A.A. Gene expression profiles during in vivo human rhinovirus infection: Insights into the host response. Am. J. Respir. Crit. Care Med. 2008, 178, 962–968. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence (5′–3′) | Purpose |
|---|---|---|
| qPCR-mouse Viperin-F | CTATCTCCTGCGACAGCT | Real time q-PCR for mouse viperin |
| qPCR-mouse Viperin-R | AAAGCCACCTTGTAATCC | |
| qPCR-Viperin-F | CAAGACCGGGGAGAATACCTG | Real time q-PCR for human viperin |
| qPCR-Viperin-R | GCGAGAATGTCCAAATACTCACC | |
| qPCR-EVA71-VP1-F: | GAGAGTTCTATA GGGGACAGT | Real time q-PCR for EVA71 viral RNA |
| qPCR-EVA71-VP1-R | AGCTGTGCTATG TGA ATTAGG AA | |
| ∆SAM 1+2- R1: | GTTGATCTTCTCCATGCGAGTGAAGTGATA | Construction of ∆SAM 1+2 |
| ∆SAM 1+2- F2: | TATCACTTCACTCGCATGGAGAAGATCAAC | |
| ∆SAM 3+4- R1: | ATTGACTTCCTCGTCGCTGGGCAGCCGCAA | Construction of ∆SAM 3+4 |
| ∆SAM 3+4- F2: | TTGCGGCTGCCCAGCGACGAGGAAGTCAAT | |
| ∆N- F: | ACTGCGGCCGCAGCCACCATGGATTACAAGGATGACGACGATAAGATGACCCCAACCAGCGTC | Construction of ∆N |
| ∆C- R: | CTAGCTCGAGGCTGTCACAGGAG | Construction of ∆C |
| ∆SAM- R1: | GACTTCCTCGTCAAAGGTGGGCAGAGGAGG | Construction of ∆SAM |
| ∆SAM- F2: | CCTCCTCTGCCCACCTTTGACGAGGAAGTC | |
| C-viperein- F: | ACTGCGGCCGCAGCCACCATGGATTACAAGGATGACGACGATAAGATGTTTGACGAGGAAGTC | Construction of C-viperin |
| N-viperin- R: | GGTGGGCAGAGGAGGGTCCTCTT | Construction of N-viperin |
| ∆N-42- F: | ACTGCGGCCGCAGCCACCATGGATTACAAGGATGACGACGATAAGATGGCTACCAAGAGGAGA | Construction of ∆N-42 |
| ∆N-50- F: | ACTGCGGCCGCAGCCACCATGGATTACAAGGATGACGACGATAAGATGCTGGTCCTGAGAGGG | Construction of ∆N-50 |
| ∆N-60- F: | ACTGCGGCCGCAGCCACCATGGATTACAAGGATGACGACGATAAGATGGAGGAGGAAGAGGAC | Construction of ∆N-60 |
| ∆N-65- F: | ACTGCGGCCGCAGCCACCATGGATTACAAGGATGACGACGATAAGATGCCTCCTCTGCCCACC | Construction of ∆N-65 |
| Si-Viperin 1 | GAGAATACCTGGGCAAGTT | Knock-down of viperin |
| Si-Viperin 2 | TAGAGTCGCTTTCAAGATA | Knock-down of viperin |
| Si-Viperin 3 | GGAGTAAGGCTGATCTGAA | Knock-down of viperin |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, C.; Zheng, C.; Sun, J.; Luo, D.; Tang, Y.; Zhang, Y.; Ke, X.; Liu, Y.; Zheng, Z.; Wang, H. Viperin Inhibits Enterovirus A71 Replication by Interacting with Viral 2C Protein. Viruses 2019, 11, 13. https://doi.org/10.3390/v11010013
Wei C, Zheng C, Sun J, Luo D, Tang Y, Zhang Y, Ke X, Liu Y, Zheng Z, Wang H. Viperin Inhibits Enterovirus A71 Replication by Interacting with Viral 2C Protein. Viruses. 2019; 11(1):13. https://doi.org/10.3390/v11010013
Chicago/Turabian StyleWei, Chunyu, Caishang Zheng, Jianhong Sun, Dan Luo, Yan Tang, Yuan Zhang, Xianliang Ke, Yan Liu, Zhenhua Zheng, and Hanzhong Wang. 2019. "Viperin Inhibits Enterovirus A71 Replication by Interacting with Viral 2C Protein" Viruses 11, no. 1: 13. https://doi.org/10.3390/v11010013
APA StyleWei, C., Zheng, C., Sun, J., Luo, D., Tang, Y., Zhang, Y., Ke, X., Liu, Y., Zheng, Z., & Wang, H. (2019). Viperin Inhibits Enterovirus A71 Replication by Interacting with Viral 2C Protein. Viruses, 11(1), 13. https://doi.org/10.3390/v11010013