Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection


{kind=link}
Abstract
1. Introduction
2. The Hurdles on the Road Towards an HIV-1 Cure
3. Molecular Determinants of HIV Latency
4. HIV-1 Integration Is Mediated by LEDGF/p75, the “Global Positioning System (GPS)” of HIV
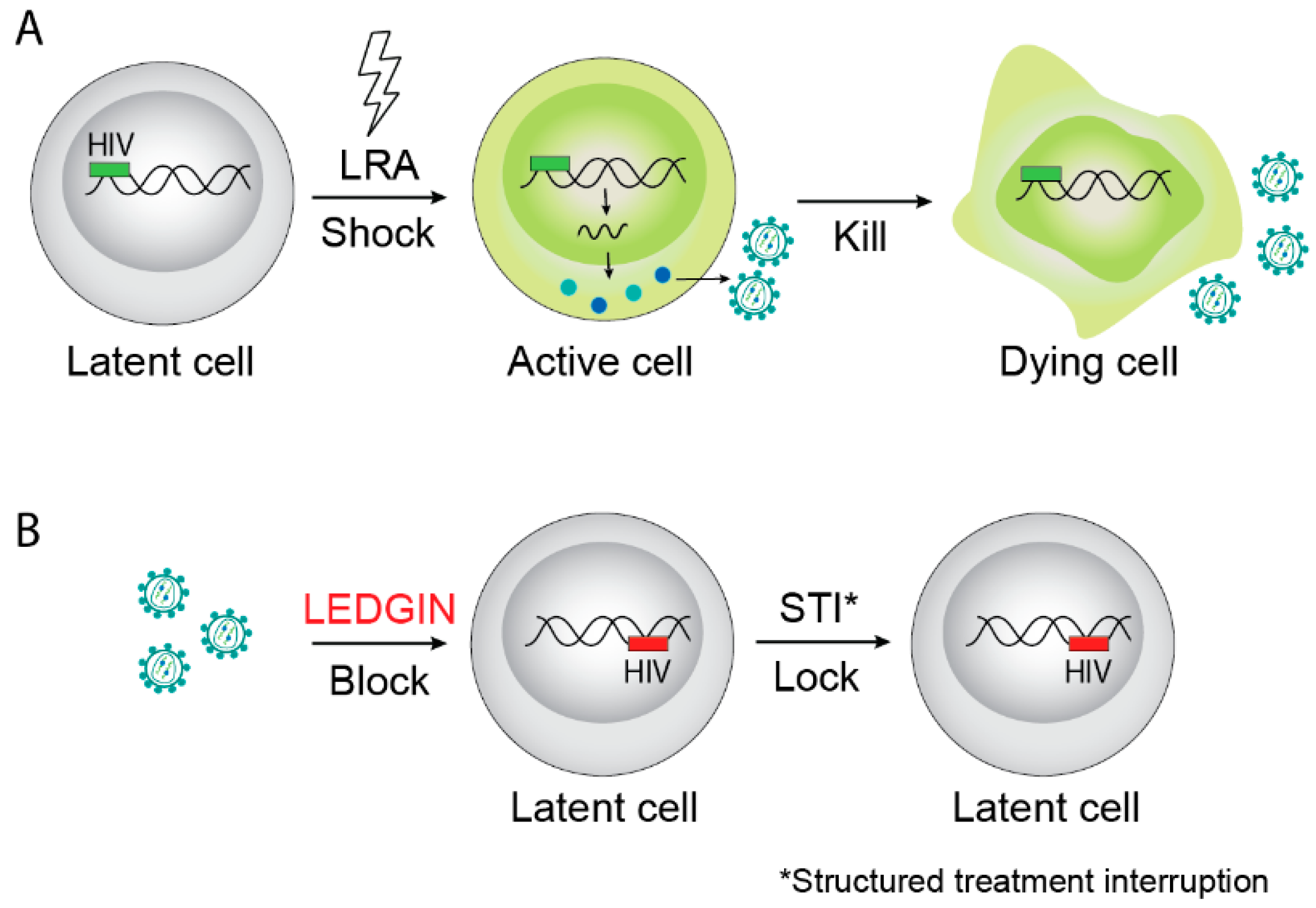
5. LEDGINs are Antivirals That Block the Interaction between HIV-1 Integrase and LEDGF/p75 and Display a Multimodal Mechanism of Action
6. LEDGF/p75 Points the Way to a Block-and-Lock Strategy for a Functional Cure of HIV Infection
7. Tat Inhibition Provides a Second Block-and-Lock Strategy
8. The Debate is Open
Funding
Conflicts of Interest
References
- UNAIDS. UNAIDS Data 2017; Joint United Nations Programme on HIV/AIDS (UNAIDS); UNAIDS Resources/UNAIDS, 2017; pp. 1–248. ISBN 978-92-9173-945-5. [Google Scholar]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.-R.; et al. International AIDS Society Global Scientific Strategy: Towards an HIV Cure 2016. Nat. Med. 2016, 22, 839. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.M.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S.; Vandekerckhove, L.; et al. Presence of an Inducible HIV-1 Latent Reservoir during Highly Active Antiretroviral Therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.; Winckelmann, A.; Palmer, S. HIV-1 Reservoirs During Suppressive Therapy. Trends Microbiol. 2016, 24, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Yukl, S.A. Tissue Reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.-Y.; Archer, J.; Kosakovsky Pond, S.L.; Chung, Y.-S.; Penugonda, S.; Chipman, J.G.; Fletcher, C.V.; et al. Persistent HIV-1 Replication Maintains the Tissue Reservoir during Therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Buzón, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 Replication and Immune Dynamics Are Affected by Raltegravir Intensification of HAART-Suppressed Subjects. Nat. Med. 2010, 16, 460. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 Replication Is Associated with Lower Antiretroviral Drug Concentrations in Lymphatic Tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef]
- Hatano, H.; Strain, M.C.; Scherzer, R.; Bacchetti, P.; Wentworth, D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Neaton, J.D.; Tracy, R.P.; et al. Increase in 2-Long Terminal Repeat Circles and Decrease in D-Dimer after Raltegravir Intensification in Patients with Treated HIV Infection: A Randomized, Placebo-Controlled Trial. J. Infect. Dis. 2013, 208, 1436–1442. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 Transcription and Latency: An Update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV Reservoirs: What, Where and How to Target Them. Nat. Rev. Microbiol. 2015, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.; Han, Y.; Zhou, Y.; Siliciano, J.; Siliciano, R.F. The Multifactorial Nature of HIV-1 Latency. Trends Mol. Med. 2004, 10, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 Latency Provides Clues for the Eradication of Long-Term Reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV Reservoir Size and Persistence Are Driven by T Cell Survival and Homeostatic Proliferation. Nat. Med. 2009, 15, 893. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 Persistence in CD4+ T Cells with Stem Cell–like Properties. Nat. Med. 2014, 20, 139. [Google Scholar] [CrossRef] [PubMed]
- Pallikkuth, S.; Sharkey, M.; Babic, D.Z.; Gupta, S.; Stone, G.W.; Fischl, M.A.; Stevenson, M.; Pahwa, S. Peripheral T Follicular Helper Cells Are the Major HIV Reservoir within Central Memory CD4 T Cells in Peripheral Blood from Chronically HIV-Infected Individuals on Combination Antiretroviral Therapy. J. Virol. 2016, 90, 2718–2728. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV Integration Sites Are Linked to Clonal Expansion and Persistence of Infected Cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of Cells with HIV Integrated into Cancer Genes Contributes to Persistent Infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. Preclinical Shock Strategies to Reactivate Latent HIV-1: An Update. Curr. Opin. HIV AIDS 2016, 11, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of Latent HIV-1 Infection in Vivo: A Proof-of-Concept Study. Lancet (Lond. Engl.) 2005, 366, 549–555. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Lai, J.; Callender, M.; Pitt, E.; Zhang, H.; Margolick, J.B.; Gallant, J.E.; Cofrancesco Joseph, J.; Moore, R.D.; Gange, S.J.; et al. Stability of the Latent Reservoir for HIV-1 in Patients Receiving Valproic Acid. J. Infect. Dis. 2007, 195, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Tremblay, C.L.; Angel, J.B.; Trottier, B.; Rouleau, D.; Baril, J.G.; Harris, M.; Trottier, S.; Singer, J.; Chomont, N.; et al. Valproic Acid in Association with Highly Active Antiretroviral Therapy for Reducing Systemic HIV-1 Reservoirs: Results from a Multicentre Randomized Clinical Study. HIV Med. 2012, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of Vorinostat Disrupts HIV-1 Latency in Patients on Antiretroviral Therapy. Nature 2012, 487, 482. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef]
- Liao, H.-K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.-J.; et al. Use of the CRISPR/Cas9 System as an Intracellular Defense against HIV-1 Infection in Human Cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.-C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-Dependent Regulation of Integrated HIV-1 Expression by Host Gene Transcriptional Readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, S0092–S8674. [Google Scholar] [CrossRef]
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The Molecular Biology of HIV Latency BT—HIV Vaccines and Cure: The Path Towards Finding an Effective Cure and Vaccine; Zhang, L., Lewin, S.R., Eds.; Springer: Singapore, 2018; pp. 187–212. [Google Scholar]
- Madison, M.N.; Okeoma, C.M. Exosomes: Implications in HIV-1 Pathogenesis. Viruses 2015, 7, 4093–4118. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-Infected Cells Stimulate Production of Pro-Inflammatory Cytokines through Trans-Activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from Uninfected Cells Activate Transcription of Latent HIV-1. J. Biol. Chem. 2017, 292, 11682–11701. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Lu, H.; Dooner, M.; Chapman, S.; Quesenberry, P.J.; Ramratnam, B. Exosomal Tat Protein Activates Latent HIV-1 in Primary, Resting CD4+ T Lymphocytes. JCI Insight 2018, 7. [Google Scholar] [CrossRef]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 Is Activated by Exosomes from Cells Infected with Either Replication-Competent or Defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Defechereux, P.; Verdin, E. The Site of HIV-1 Integration in the Human Genome Determines Basal Transcriptional Activity and Response to Tat Transactivation. EMBO J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef]
- Maxfield, L.F.; Fraize, C.D.; Coffin, J.M. Relationship between Retroviral DNA-Integration-Site Selection and Host Cell Transcription. Proc. Natl. Acad. Sci. USA 2005, 102, 1436–1441. [Google Scholar] [CrossRef]
- Felice, B.; Cattoglio, C.; Cittaro, D.; Testa, A.; Miccio, A.; Ferrari, G.; Luzi, L.; Recchia, A.; Mavilio, F. Transcription Factor Binding Sites Are Genetic Determinants of Retroviral Integration in the Human Genome. PLoS ONE 2009, 4, e4571. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L. Nuclear Architecture Dictates HIV-1 Integration Site Selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Arosio, D.; Terreni, M.; Cereseto, A. HIV-1 Pre-Integration Complexes Selectively Target Decondensed Chromatin in the Nuclear Periphery. PLoS ONE 2008, 3, e2413. [Google Scholar] [CrossRef] [PubMed]
- Dieudonné, M.; Maiuri, P.; Biancotto, C.; Knezevich, A.; Kula, A.; Lusic, M.; Marcello, A. Transcriptional Competence of the Integrated HIV-1 Provirus at the Nuclear Periphery. EMBO J. 2009, 28, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.; Sankale, J.-L.; Meloni, S.T.; Sarr, A.D.; Mboup, S.; Kanki, P. Genomic Sites of Human Immunodeficiency Virus Type 2 (HIV-2) Integration: Similarities to HIV-1 In Vitro and Possible Differences In Vivo. J. Virol. 2006, 80, 7316–7321. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Hong, B.-K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct Genomic Integration of MLV and SIV Vectors in Primate Hematopoietic Stem and Progenitor Cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.J.; Peña, Á.; Vallejo, F.G. A Genomic and Bioinformatics Analysis of the Integration of HIV in Peripheral Blood Mononuclear Cells. AIDS Res. Hum. Retroviruses 2010, 27, 547–555. [Google Scholar] [CrossRef]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host Factors for Retroviral Integration Site Selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 Integrase Forms Stable Tetramers and Associates with LEDGF/P75 Protein in Human Cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef]
- Cherepanov, P. LEDGF/P75 Interacts with Divergent Lentiviral Integrases and Modulates Their Enzymatic Activity in Vitro. Nucleic Acids Res. 2007, 35, 113–124. [Google Scholar] [CrossRef]
- Chylack, L.T.; Fu, L.; Mancini, R.; Martin-Rehrmann, M.D.; Saunders, A.J.; Konopka, G.; Tian, D.; Hedley-Whyte, E.T.; Folkerth, R.D.; Goldstein, L.E. Lens Epithelium-Derived Growth Factor (LEDGF/P75) Expression in Fetal and Adult Human Brain. Exp. Eye Res. 2004, 79, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Kimura, A.; Chylack, L.T.; Shinohara, T. Lens Epithelium-Derived Growth Factor (LEDGF/P75) and P52 Are Derived from a Single Gene by Alternative Splicing. Gene 2000, 242, 265–273. [Google Scholar] [CrossRef]
- Sutherland, H.G.; Newton, K.; Brownstein, D.G.; Holmes, M.C.; Kress, C.; Semple, C.A.; Bickmore, W.A. Disruption of Ledgf/Psip1 Results in Perinatal Mortality and Homeotic Skeletal Transformations. Mol. Cell. Biol. 2006, 26, 7201–7210. [Google Scholar] [CrossRef] [PubMed]
- Ochs, R.L.; Muro, Y.; Si, Y.; Ge, H.; Chan, E.K.L.; Tan, E.M. Autoantibodies to DFS 70 Kd/Transcription Coactivator P75 in Atopic Dermatitis and Other Conditions. J. Allergy Clin. Immunol. 2000, 105, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Cajigas-Du Ross, C.K.; Rios-Colon, L.; Mediavilla-Varela, M.; Daniels-Wells, T.R.; Leoh, L.S.; Rojas, H.; Banerjee, H.; Martinez, S.R.; Acevedo-Martinez, S.; et al. LEDGF/P75 Overexpression Attenuates Oxidative Stress-Induced Necrosis and Upregulates the Oxidoreductase ERP57/PDIA3/GRP58 in Prostate Cancer. PLoS ONE 2016, 11, e0146549. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Eidahl, J.O.; Crowe, B.L.; North, J.A.; McKee, C.J.; Shkriabai, N.; Feng, L.; Plumb, M.; Graham, R.L.; Gorelick, R.J.; Hess, S.; et al. Structural Basis for High-Affinity Binding of LEDGF PWWP to Mononucleosomes. Nucleic Acids Res. 2013, 41, 3924–3936. [Google Scholar] [CrossRef]
- Van Nuland, R.; van Schaik, F.M.; Simonis, M.; van Heesch, S.; Cuppen, E.; Boelens, R.; Timmers, H.M.; van Ingen, H. Nucleosomal DNA Binding Drives the Recognition of H3K36-Methylated Nucleosomes by the PSIP1-PWWP Domain. Epigenetics Chromatin 2013, 6, 12. [Google Scholar] [CrossRef]
- Ge, H.; Si, Y.; Roeder, R.G.; Bjorklund, S.; Kim, Y.; Burke, T.; Kadonaga, J.; Burke, T.; Kadonaga, J.; Burley, S.; et al. Isolation of CDNAs Encoding Novel Transcription Coactivators P52 and P75 Reveals an Alternate Regulatory Mechanism of Transcriptional Activation. EMBO J. 1998, 17, 6723–6729. [Google Scholar] [CrossRef]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an Evolutionarily Conserved Domain in Human Lens Epithelium-Derived Growth Factor/Transcriptional Co-Activator P75 (LEDGF/P75) That Binds HIV-1 Integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef]
- Cermakova, K.; Weydert, C.; Christ, F.; De Rijck, J.; Debyser, Z. Lessons Learned: HIV Points the Way Towards Precision Treatment of Mixed-Lineage Leukemia. Trends Pharmacol. Sci. 2016, 37, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Ambrosio, A.L.B.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural Basis for the Recognition between HIV-1 Integrase and Transcriptional Coactivator P75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Shun, M.-C.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. A Novel Co-Crystal Structure Affords the Design of Gain-of-Function Lentiviral Integrase Mutants in the Presence of Modified PSIP1/LEDGF/P75. PLoS Pathog. 2009, 5, e1000259. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A Role for LEDGF/P75 in Targeting HIV DNA Integration. Nat. Med. 2005, 11, 1287. [Google Scholar] [CrossRef]
- Shun, M.-C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/P75 Functions Downstream from Preintegration Complex Formation to Effect Gene-Specific HIV-1 Integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An Essential Role for LEDGF/P75 in HIV Integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and Stable Knockdown of the Integrase Cofactor LEDGF/P75 Reveals Its Role in the Replication Cycle of Human Immunodeficiency Virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, R.; Vets, S.; De Rijck, J.; Malani, N.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. HRP-2 Determines HIV-1 Integration Site Selection in LEDGF/P75 Depleted Cells. Retrovirology 2012, 9, 84. [Google Scholar] [CrossRef]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The Interaction of LEDGF/P75 with Integrase Is Lentivirus-Specific and Promotes DNA Binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef]
- Llano, M.; Delgado, S.; Vanegas, M.; Poeschla, E.M. Lens Epithelium-Derived Growth Factor/P75 Prevents Proteasomal Degradation of HIV-1 Integrase. J. Biol. Chem. 2004, 279, 55570–55577. [Google Scholar] [CrossRef]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/P75-Independent HIV-1 Replication Demonstrates a Role for HRP-2 and Remains Sensitive to Inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; Vandekerckhove, L.; Gijsbers, R.; Hombrouck, A.; Hendrix, J.; Vercammen, J.; Engelborghs, Y.; Christ, F.; Debyser, Z. Overexpression of the Lens Epithelium-Derived Growth Factor/P75 Integrase Binding Domain Inhibits Human Immunodeficiency Virus Replication. J. Virol. 2006, 80, 11498–11509. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational Design of Small-Molecule Inhibitors of the LEDGF/P75-Integrase Interaction and HIV Replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Demeulemeester, J.; Chaltin, P.; Marchand, A.; De Maeyer, M.; Debyser, Z.; Christ, F. LEDGINs, Non-Catalytic Site Inhibitors of HIV-1 Integrase: A Patent Review (2006–2014). Expert Opin. Ther. Pat. 2014, 24, 609–632. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Shaw, S.; Demeulemeester, J.; Desimmie, B.A.; Marchand, A.; Butler, S.; Smets, W.; Chaltin, P.; Westby, M.; Debyser, Z.; et al. Small-Molecule Inhibitors of the LEDGF/P75 Binding Site of Integrase Block HIV Replication and Modulate Integrase Multimerization. Antimicrob. Agents Chemother. 2012, 56, 4365–4374. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.J.; Engelman, A.; et al. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; De Rijck, J.; et al. LEDGINs Inhibit Late Stage HIV-1 Replication by Modulating Integrase Multimerization in the Virions. Retrovirology 2013, 10, 57. [Google Scholar] [CrossRef]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric Integrase Inhibitor Potency Is Determined through the Inhibition of HIV-1 Particle Maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PLoS ONE 2013, 8, e74163. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.-M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual Inhibition of HIV-1 Replication by Integrase-LEDGF Allosteric Inhibitors Is Predominant at the Post-Integration Stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-Mediated Inhibition of Integrase–LEDGF/P75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Tojal Da Silva, I.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct Chromatin Functional States Correlate with HIV Latency Reversal in Infected Primary CD4+ T Cells. eLife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Chen, H.-C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position Effects Influence HIV Latency Reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Zorita, E.; Filion, G.J. Using Barcoded HIV Ensembles (B-HIVE) for Single Provirus Transcriptomics. Curr. Protoc. Mol. Biol. 2018, 122, e56. [Google Scholar] [CrossRef] [PubMed]
- Puray-Chavez, M.; Tedbury, P.R.; Huber, A.D.; Ukah, O.B.; Yapo, V.; Liu, D.; Ji, J.; Wolf, J.J.; Engelman, A.N.; Sarafianos, S.G. Multiplex Single-Cell Visualization of Nucleic Acids and Protein during HIV Infection. Nat. Commun. 2017, 8, 1882. [Google Scholar] [CrossRef] [PubMed]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR Nucleosome Positioning by the BAF Complex Is Required for HIV Latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef]
- Gérard, A.; Ségéral, E.; Naughtin, M.; Abdouni, A.; Charmeteau, B.; Cheynier, R.; Rain, J.-C.; Emiliani, S. The Integrase Cofactor LEDGF/P75 Associates with Iws1 and Spt6 for Postintegration Silencing of HIV-1 Gene Expression in Latently Infected Cells. Cell Host Microbe 2015, 17, 107–117. [Google Scholar] [CrossRef]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.M.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Cure. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6, e00465-15. [Google Scholar] [CrossRef]
- Mousseau, G.; Mediouni, S.; Valente, S.T. Targeting HIV Transcription: The Quest for a Functional Cure. Curr. Top. Microbiol. Immunol. 2015, 389, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Hocqueloux, L.; Avettand-Fènoël, V.; Jacquot, S.; Prazuck, T.; Legac, E.; Mélard, A.; Niang, M.; Mille, C.; Le Moal, G.; Viard, J.-P.; et al. Long-Term Antiretroviral Therapy Initiated during Primary HIV-1 Infection Is Key to Achieving Both Low HIV Reservoirs and Normal T Cell Counts. J. Antimicrob. Chemother. 2013, 68, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Malatinkova, E.; De Spiegelaere, W.; Bonczkowski, P.; Kiselinova, M.; Vervisch, K.; Trypsteen, W.; Johnson, M.; Verhofstede, C.; de Looze, D.; Murray, C.; et al. Impact of a Decade of Successful Antiretroviral Therapy Initiated at HIV-1 Seroconversion on Blood and Rectal Reservoirs. eLife 2015, 4, e09115. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Martin-Gayo, E.; Pereyra, F.; Ouyang, Z.; Sun, H.; Li, J.Z.; Piovoso, M.; Shaw, A.; Dalmau, J.; Zangger, N.; et al. Long-Term Antiretroviral Treatment Initiated at Primary HIV-1 Infection Affects the Size, Composition, and Decay Kinetics of the Reservoir of HIV-1-Infected CD4 T Cells. J. Virol. 2014, 88, 10056–10065. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debyser, Z.; Vansant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 2019, 11, 12. https://doi.org/10.3390/v11010012
Debyser Z, Vansant G, Bruggemans A, Janssens J, Christ F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses. 2019; 11(1):12. https://doi.org/10.3390/v11010012
Chicago/Turabian StyleDebyser, Zeger, Gerlinde Vansant, Anne Bruggemans, Julie Janssens, and Frauke Christ. 2019. "Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection" Viruses 11, no. 1: 12. https://doi.org/10.3390/v11010012
APA StyleDebyser, Z., Vansant, G., Bruggemans, A., Janssens, J., & Christ, F. (2019). Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses, 11(1), 12. https://doi.org/10.3390/v11010012