The Hard Way towards an Antibody-Based HIV-1 Env Vaccine: Lessons from Other Viruses



Abstract
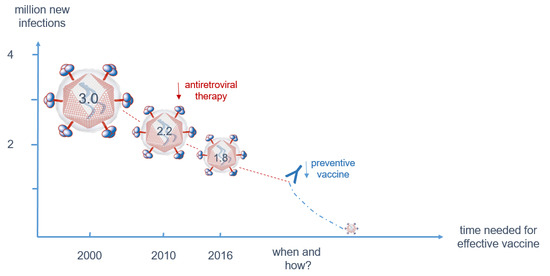
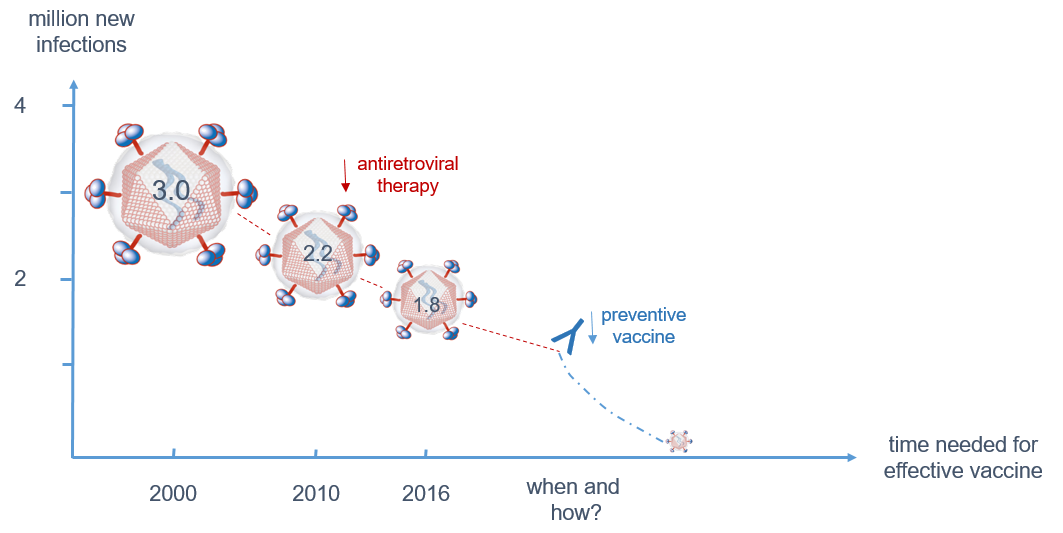{kind=link}
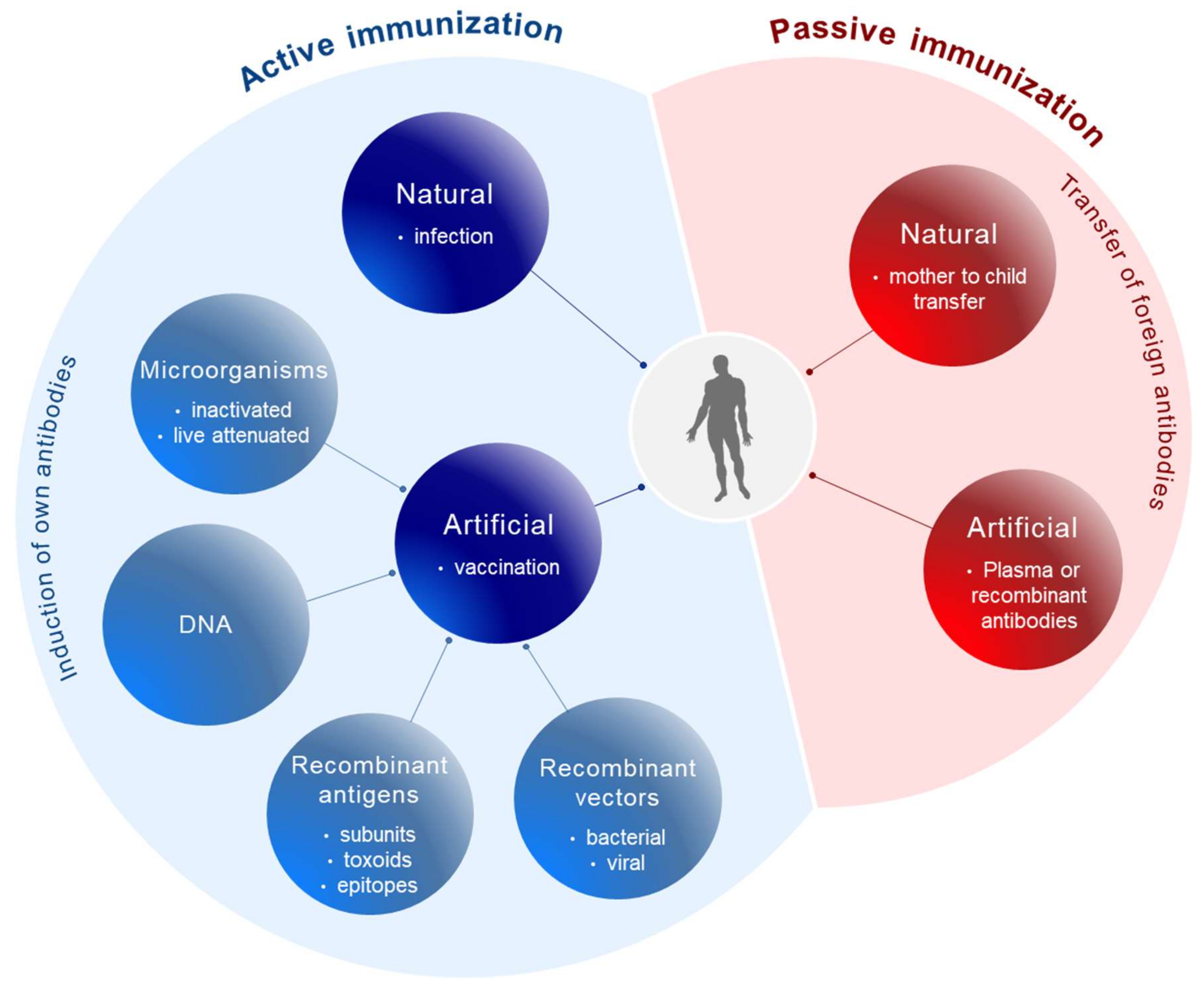{kind=link}
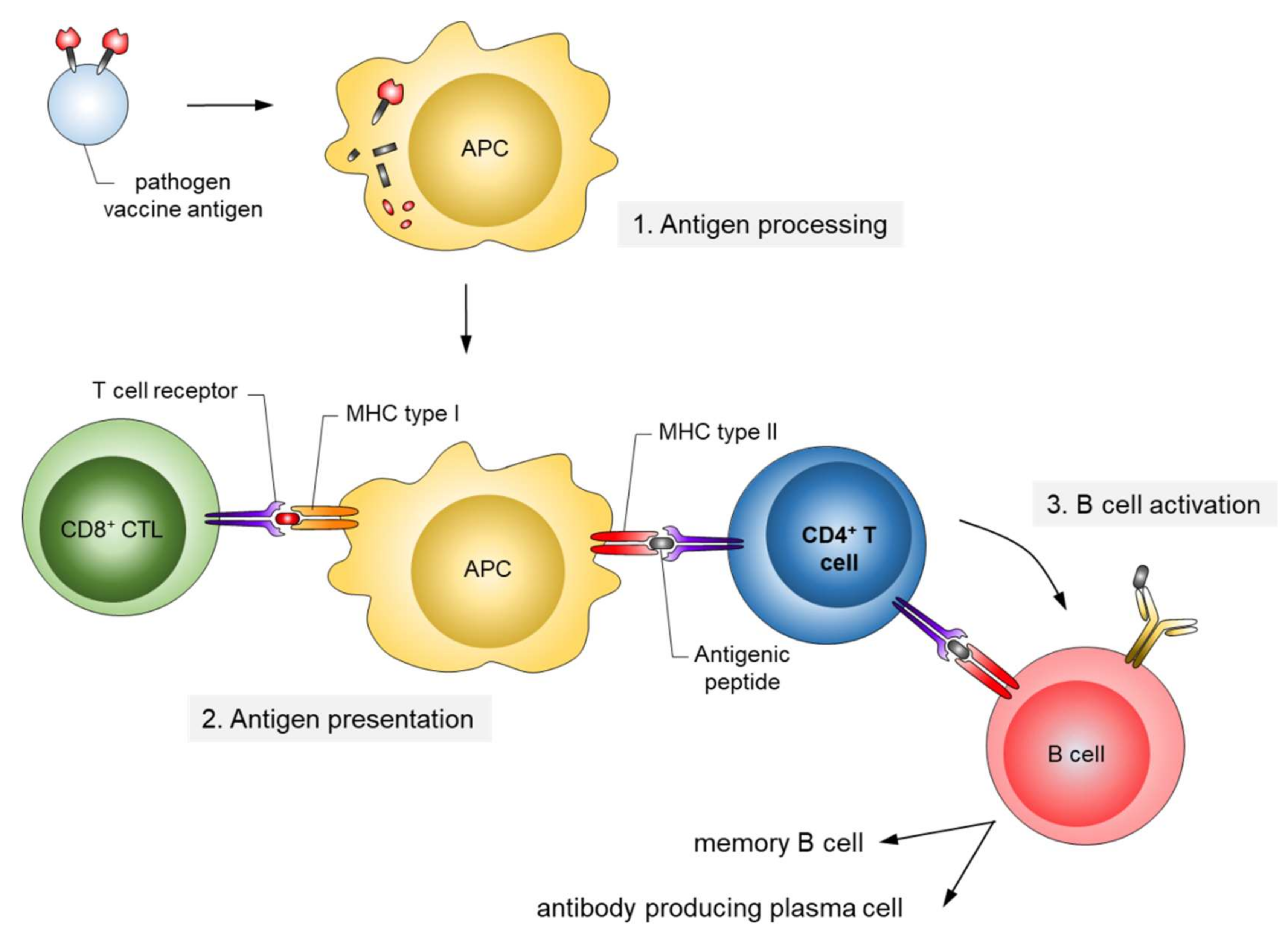{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. From Whole Pathogen and Subunit Vaccines to Epitope Vaccines
3. HIV-1 Vaccine Development
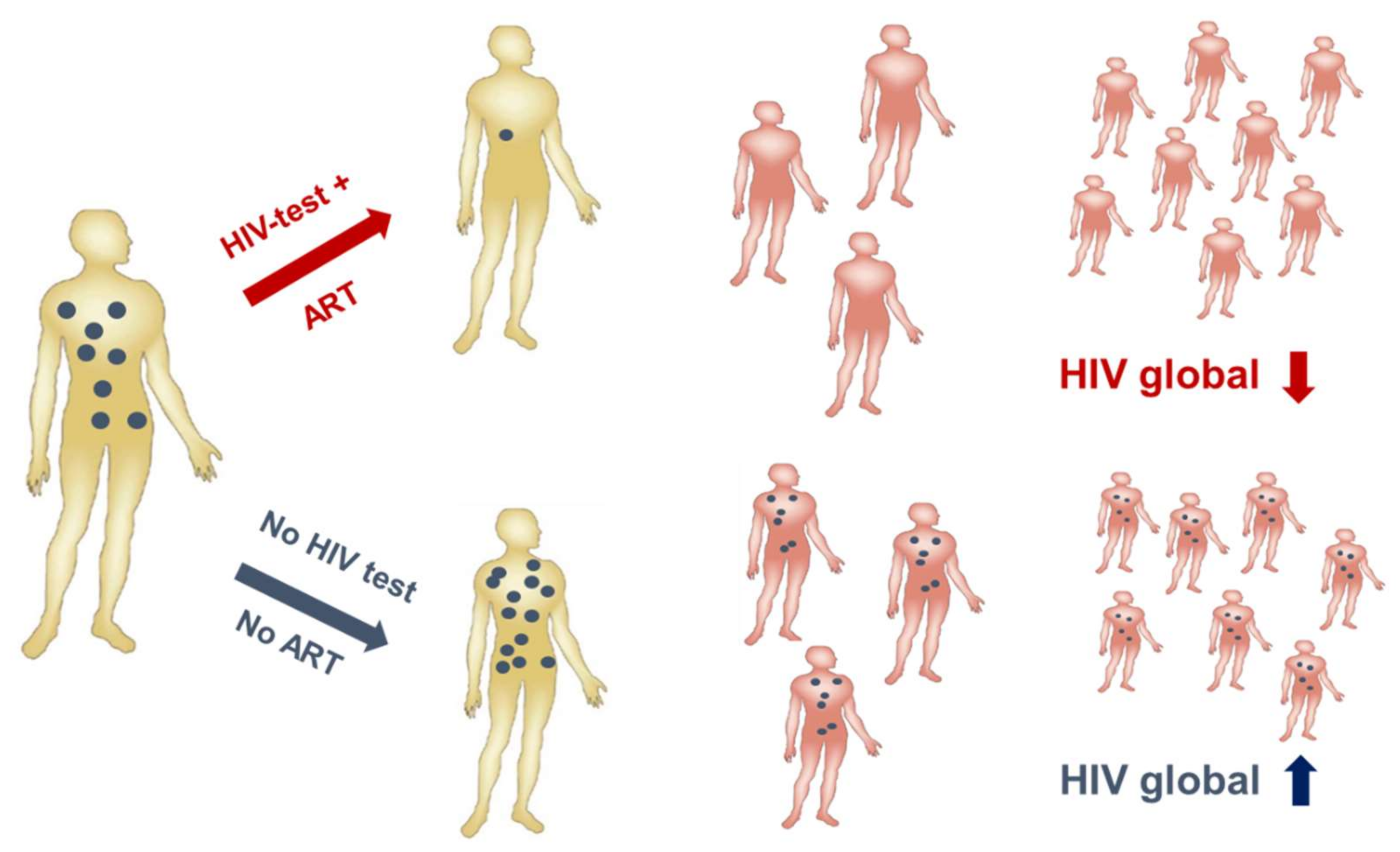
3.1. Despite Effective Anti-Retroviral Treatment (ART), a Vaccine is Still Needed to Contain the Epidemic
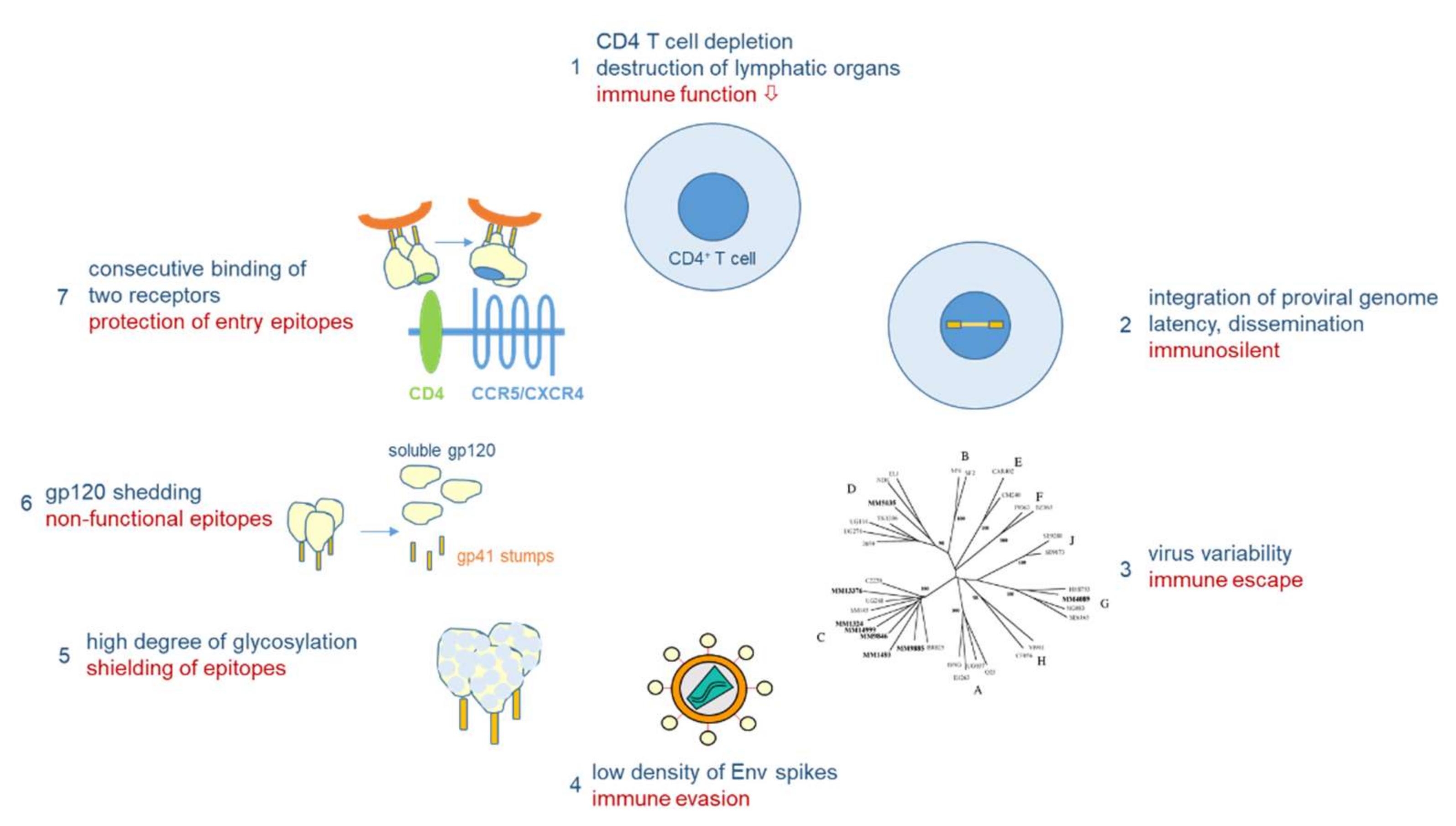
3.2. The Bag of Tricks of HIV-1 to Evade the Immune System also Hinders Vaccine Development
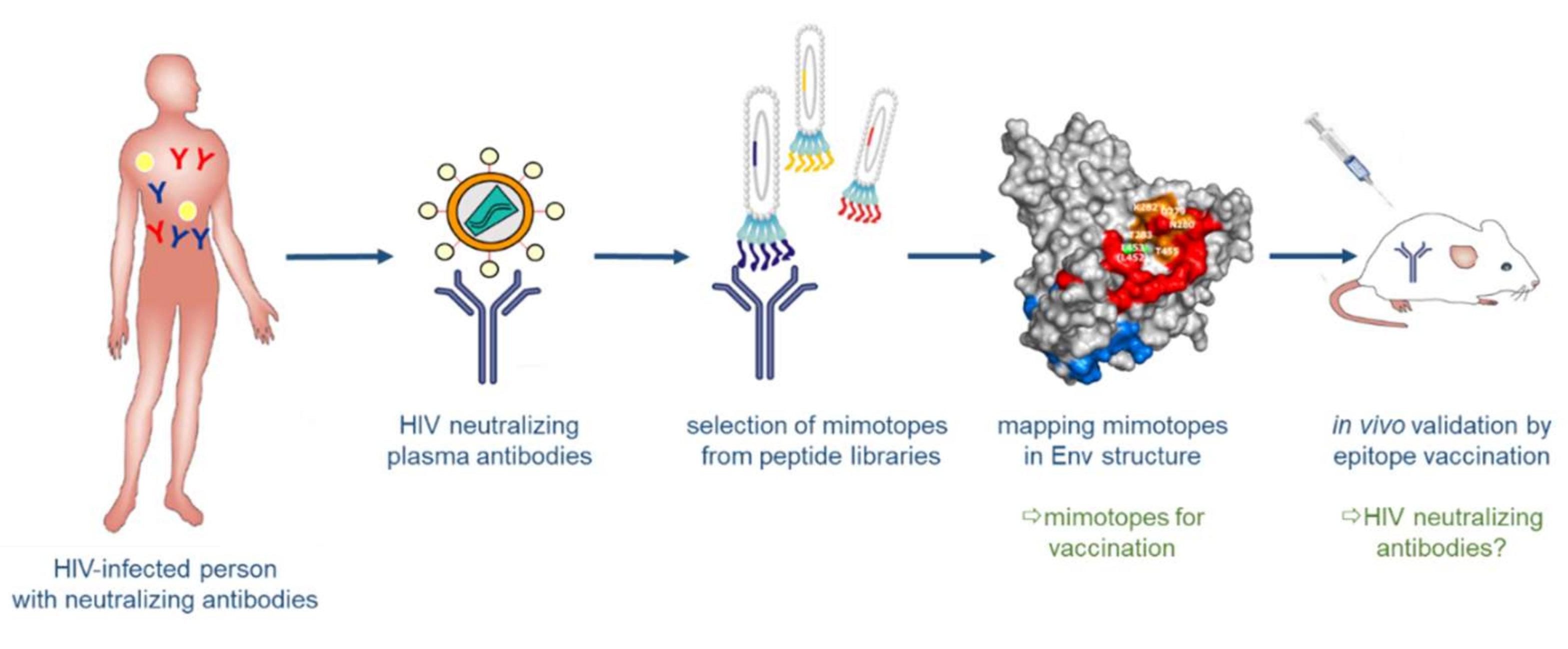
3.3. Structure-Based Reverse Vaccinology and Epitope Vaccines for HIV-1
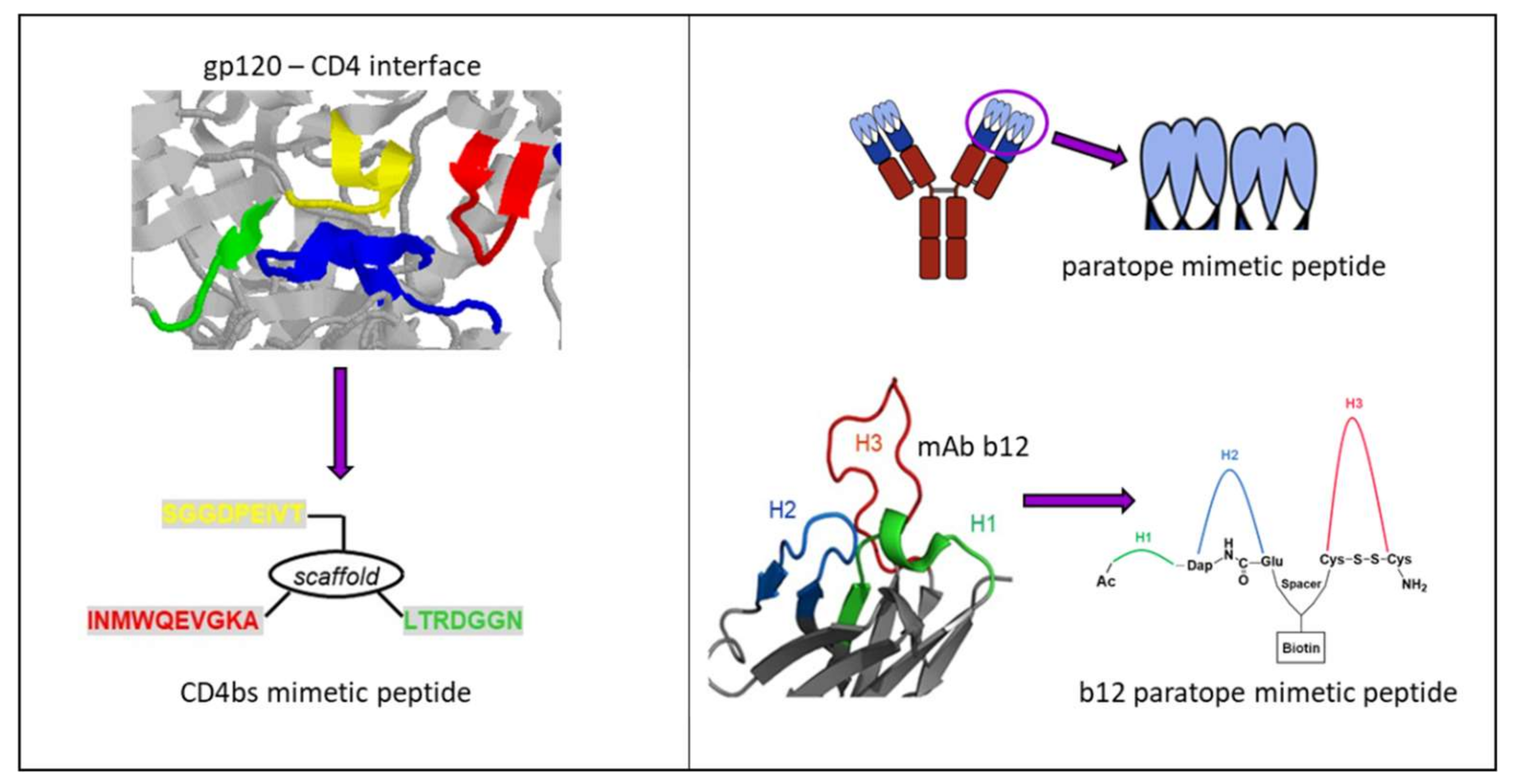
3.3.1. Synthetic Peptide Mimics as Vaccine Candidates
3.3.2. Two Promising gp41 Epitope Vaccine Candidates
3.4. Vectored Expression of bnAbs
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- WHO. Smallpox in the Post-Eradication Era. Available online: http://www.who.int/wer/2016/wer9120.pdf?ua=1 (accessed on 23 February 2018).
- Greenwood, B. The contribution of vaccination to global health: Past, present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130433. [Google Scholar] [CrossRef] [PubMed]
- Wojda, T.R.; Valenza, P.L.; Cornejo, K.; McGinley, T.; Galwankar, S.C.; Kelkar, D.; Sharpe, R.P.; Papadimos, T.J.; Stawicki, S.P. The Ebola Outbreak of 2014–2015: From Coordinated Multilateral Action to Effective Disease Containment, Vaccine Development, and Beyond. J. Glob. Infect. Dis. 2015, 7, 127–138. [Google Scholar] [PubMed]
- CEPI. New Vaccines for a Safer World. Available online: http://cepi.net/sites/default/files/CEPI%20booklet%20final_0.pdf (accessed on 23 February 2018).
- Diamond, B. Global polio campaign doomed to fail, experts warn. Nat. Med. 2005, 11, 1260. [Google Scholar] [CrossRef] [PubMed]
- Willyard, C. Polio eradication campaign copes with unusual outbreak. Nat. Med. 2007, 13, 1394. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, J.J.; Eley, B.S. BCG Vaccination in HIV-Infected Children. Tuberc. Res. Treat. 2011, 2011, 712736. [Google Scholar] [CrossRef] [PubMed]
- He, Y. Immunogenicity of SARS-CoV: The receptor-binding domain of S protein is a major target of neutralizing antibodies. Adv. Exp. Med. Biol. 2006, 581, 539–542. [Google Scholar] [PubMed]
- Belshe, R.B. The need for quadrivalent vaccine against seasonal influenza. Vaccine 2010, 28 (Suppl. 4), D45–D53. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; Altenburg, A.F.; Rimmelzwaan, G.F. Universal influenza vaccines, science fiction or soon reality? Expert Rev. Vaccines 2015, 14, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Gerdil, C. The annual production cycle for influenza vaccine. Vaccine 2003, 21, 1776–1779. [Google Scholar] [CrossRef]
- Veljkovic, V.; Paessler, S.; Glisic, S.; Prljic, J.; Perovic, V.R.; Veljkovic, N.; Scotch, M. Evolution of 2014/15 H3N2 Influenza Viruses Circulating in US: Consequences for Vaccine Effectiveness and Possible New Pandemic. Front. Microbiol. 2015, 6, 1456. [Google Scholar] [CrossRef] [PubMed]
- Perovic, V.R.; Muller, C.P.; Niman, H.L.; Veljkovic, N.; Dietrich, U.; Tosic, D.D.; Glisic, S.; Veljkovic, V. Novel phylogenetic algorithm to monitor human tropism in Egyptian H5N1-HPAIV reveals evolution toward efficient human-to-human transmission. PLoS ONE 2013, 8, e61572. [Google Scholar] [CrossRef] [PubMed]
- Schmier, S.; Mostafa, A.; Haarmann, T.; Bannert, N.; Ziebuhr, J.; Veljkovic, V.; Dietrich, U.; Pleschka, S. In Silico Prediction and Experimental Confirmation of HA Residues Conferring Enhanced Human Receptor Specificity of H5N1 Influenza A Viruses. Sci. Rep. 2015, 5, 11434. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Giles, B.M.; Bissel, S.J.; Dealmeida, D.R.; Wiley, C.A.; Ross, T.M. Antibody breadth and protective efficacy are increased by vaccination with computationally optimized hemagglutinin but not with polyvalent hemagglutinin-based H5N1 virus-like particle vaccines. Clin. Vaccine Immunol. 2012, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Giles, B.M.; Ross, T.M. Computationally optimized antigens to overcome influenza viral diversity. Expert Rev. Vaccines 2012, 11, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Throsby, M.; van den Brink, E.; Jongeneelen, M.; Poon, L.L.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE 2008, 3, e3942. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.D.; Bajic, G.; Ferdman, J.; Suphaphiphat, P.; Settembre, E.C.; Moody, M.A.; Schmidt, A.G.; Harrison, S.C. Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc. Natl. Acad. Sci. USA 2018, 115, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.G.; Therkelsen, M.D.; Stewart, S.; Kepler, T.B.; Liao, H.X.; Moody, M.A.; Haynes, B.F.; Harrison, S.C. Viral receptor-binding site antibodies with diverse germline origins. Cell 2015, 161, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Poignard, P.; Stanfield, R.L.; Wilson, I.A. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 2012, 337, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science 2012, 337, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Friesen, R.H.; Lee, P.S.; Stoop, E.J.; Hoffman, R.M.; Ekiert, D.C.; Bhabha, G.; Yu, W.; Juraszek, J.; Koudstaal, W.; Jongeneelen, M.; et al. A common solution to group 2 influenza virus neutralization. Proc. Natl. Acad. Sci. USA 2014, 111, 445–450. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; Nieuwkoop, N.J.; van der Klis, F.R.M.; Koopmans, M.P.G.; Krammer, F.; Rimmelzwaan, G.F. Primary Human Influenza B Virus Infection Induces Cross-Lineage Hemagglutinin Stalk-Specific Antibodies Mediating Antibody-Dependent Cellular Cytoxicity. J. Infect. Dis. 2017, 217, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.; van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Lu, L.; Liu, Q.; Xu, W.; Du, L. Receptor-binding domains of spike proteins of emerging or re-emerging viruses as targets for development of antiviral vaccines. Emerg. Microbes Infect. 2012, 1, e13. [Google Scholar] [CrossRef] [PubMed]
- Negahdaripour, M.; Nezafat, N.; Eslami, M.; Ghoshoon, M.B.; Shoolian, E.; Najafipour, S.; Morowvat, M.H.; Dehshahri, A.; Erfani, N.; Ghasemi, Y. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect. Genet. Evol. 2018, 58, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Sher, G.; Zhi, D.; Zhang, S. DRREP: Deep ridge regressed epitope predictor. BMC Genomics 2017, 18 (Suppl. 6), 676. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, N.C.; Kohlbacher, O. Towards in silico design of epitope-based vaccines. Expert Opin. Drug Discov. 2009, 4, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Oyarzun, P.; Kobe, B. Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum. Vaccine Immunother. 2016, 12, 763–767. [Google Scholar] [CrossRef] [PubMed]
- White, M.T.; Verity, R.; Griffin, J.T.; Asante, K.P.; Owusu-Agyei, S.; Greenwood, B.; Drakeley, C.; Gesase, S.; Lusingu, J.; Ansong, D.; et al. Immunogenicity of the RTS,S/AS01 malaria vaccine and implications for duration of vaccine efficacy: Secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect. Dis. 2015, 15, 1450–1458. [Google Scholar] [CrossRef]
- Gosling, R.; von Seidlein, L. The Future of the RTS,S/AS01 Malaria Vaccine: An Alternative Development Plan. PLoS Med. 2016, 13, e1001994. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, E.; Liu, H.; Ben-Yedidia, T.; Hassin, S.; Visontai, I.; Norley, S.; Frijlink, H.W.; Hak, E. Evaluating the immunogenicity and safety of a BiondVax-developed universal influenza vaccine (Multimeric-001) either as a standalone vaccine or as a primer to H5N1 influenza vaccine: Phase IIb study protocol. Medicine (Baltimore) 2017, 96, e6339. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS 90-90-90—An Ambitious Treatment Target to Help End the AIDS Epidemic. Available online: http://www.unaids.org/en/resources/documents/2017/90-90-90 (accessed on 1 February 2018).
- UNAIDS. UNAIDS Data 2017. Available online: http://www.unaids.org (accessed on 1 February 2018).
- Burns, D.N.; DeGruttola, V.; Pilcher, C.D.; Kretzschmar, M.; Gordon, C.M.; Flanagan, E.H.; Duncombe, C.; Cohen, M.S. Toward an endgame: Finding and engaging people unaware of their HIV-1 infection in treatment and prevention. AIDS Res. Hum. Retrovir. 2014, 30, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, U. Origin and current aspects of the HIV pandemic. Pharmakon 2014, 2, 244–249. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A. Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses 2017, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Douek, D.C. The mucosal barrier and immune activation in HIV pathogenesis. Curr. Opin. HIV AIDS 2008, 3, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A. HIV and AIDS in relation to other pandemics. Among the viruses plaguing humans, HIV is a recent acquisition. Its outstanding success as an infection poses immense scientific challenges to human health and raises the question “What comes nest?”. EMBO Rep. 2003, 4, S10–S14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geiss, Y.; Dietrich, U. Catch Me If You Can—The Race Between HIV and Neutralizing Antibodies. AIDS Rev. 2015, 17, 107–113. [Google Scholar] [PubMed]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Diedrich, J.K.; Kulp, D.W.; Pauthner, M.; He, L.; Park, S.R.; Sok, D.; Su, C.Y.; Delahunty, C.M.; Menis, S.; et al. Global site-specific N-glycosylation analysis of HIV envelope glycoprotein. Nat. Commun. 2017, 8, 14954. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.K.; Spellman, M.W.; Riddle, L.; Harris, R.J.; Thomas, J.N.; Gregory, T.J. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 1990, 265, 10373–10382. [Google Scholar] [PubMed]
- Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J.D.; Grise, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Swanstrom, R. The evolution of HIV-1 entry phenotypes as a guide to changing target cells. J. Leukoc. Biol. 2018, 103, 421–431. [Google Scholar] [CrossRef] [PubMed]
- De Taeye, S.W.; Moore, J.P.; Sanders, R.W. HIV-1 Envelope Trimer Design and Immunization Strategies to Induce Broadly Neutralizing Antibodies. Trends Immunol. 2016, 37, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Pena, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Moore, J.P. Native-like Env trimers as a platform for HIV-1 vaccine design. Immunol. Rev. 2017, 275, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Vesanen, M.; Schuelke, N.; Master, A.; Schiffner, L.; Kalyanaraman, R.; Paluch, M.; Berkhout, B.; Maddon, P.J.; Olson, W.C.; et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 2002, 76, 8875–8889. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J.; LaBranche, C.C.; Ketas, T.J.; Ozorowski, G.; Cupo, A.; Pugach, P.; Ringe, R.P.; Golabek, M.; van Gils, M.J.; Guttman, M.; et al. Sequential and Simultaneous Immunization of Rabbits with HIV-1 Envelope Glycoprotein SOSIP.664 Trimers from Clades A, B and C. PLoS Pathog. 2016, 12, e1005864. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 VACCINES. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349, aac4223. [Google Scholar] [CrossRef] [PubMed]
- Forsman, A.; Beirnaert, E.; Aasa-Chapman, M.M.; Hoorelbeke, B.; Hijazi, K.; Koh, W.; Tack, V.; Szynol, A.; Kelly, C.; McKnight, A.; et al. Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J. Virol. 2008, 82, 12069–12081. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.E.; Quigley, A.F.; Strokappe, N.M.; Bulmer-Thomas, B.; Seaman, M.S.; Mortier, D.; Rutten, L.; Chander, N.; Edwards, C.J.; Ketteler, R.; et al. Potent and broad neutralization of HIV-1 by a llama antibody elicited by immunization. J. Exp. Med. 2012, 209, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Kalusche, S.; Torres, J.L.; Stanfield, R.L.; Danquah, W.; Khazanehdari, K.; von Briesen, H.; Geertsma, E.R.; Wilson, I.A.; Wernery, U.; et al. Selection of nanobodies with broad neutralizing potential against primary HIV-1 strains using soluble subtype C gp140 envelope trimers. Sci. Rep. 2017, 7, 8390. [Google Scholar] [CrossRef] [PubMed]
- Sok, D.; Le, K.M.; Vadnais, M.; Saye-Francisco, K.L.; Jardine, J.G.; Torres, J.L.; Berndsen, Z.T.; Kong, L.; Stanfield, R.; Ruiz, J.; et al. Rapid elicitation of broadly neutralizing antibodies to HIV by immunization in cows. Nature 2017, 548, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Pejchal, R.; Doores, K.J.; Walker, L.M.; Khayat, R.; Huang, P.S.; Wang, S.K.; Stanfield, R.L.; Julien, J.P.; Ramos, A.; Crispin, M.; et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 2011, 334, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Simek, M.D.; Rida, W.; Priddy, F.H.; Pung, P.; Carrow, E.; Laufer, D.S.; Lehrman, J.K.; Boaz, M.; Tarragona-Fiol, T.; Miiro, G.; et al. Human immunodeficiency virus type 1 elite neutralizers: Individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J. Virol. 2009, 83, 7337–7348. [Google Scholar] [CrossRef] [PubMed]
- Eroshkin, A.M.; LeBlanc, A.; Weekes, D.; Post, K.; Li, Z.; Rajput, A.; Butera, S.T.; Burton, D.R.; Godzik, A. bNAber: Database of broadly neutralizing HIV antibodies. Nucleic Acids Res. 2014, 42, D1133–D1139. [Google Scholar] [CrossRef] [PubMed]
- Sliepen, K.; Sanders, R.W. HIV-1 envelope glycoprotein immunogens to induce broadly neutralizing antibodies. Expert Rev. Vaccines 2016, 15, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.B.; West, A.P., Jr. Antibody gene transfer for HIV immunoprophylaxis. Nat. Immunol. 2013, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Klein, F.; Halper-Stromberg, A.; Horwitz, J.A.; Gruell, H.; Scheid, J.F.; Bournazos, S.; Mouquet, H.; Spatz, L.A.; Diskin, R.; Abadir, A.; et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 2012, 492, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Moldt, B.; Rakasz, E.G.; Schultz, N.; Chan-Hui, P.Y.; Swiderek, K.; Weisgrau, K.L.; Piaskowski, S.M.; Bergman, Z.; Watkins, D.I.; Poignard, P.; et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18921–18925. [Google Scholar] [CrossRef] [PubMed]
- Pietzsch, J.; Gruell, H.; Bournazos, S.; Donovan, B.M.; Klein, F.; Diskin, R.; Seaman, M.S.; Bjorkman, P.J.; Ravetch, J.V.; Ploss, A.; et al. A mouse model for HIV-1 entry. Proc. Natl. Acad. Sci. USA 2012, 109, 15859–15864. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Wang, L.; Joyce, M.G.; Yang, Z.Y.; Balazs, A.B.; Cheng, C.; Ko, S.Y.; Kong, W.P.; Rudicell, R.S.; Georgiev, I.S.; et al. Broadly Neutralizing Human Immunodeficiency Virus Type 1 Antibody Gene Transfer Protects Nonhuman Primates from Mucosal Simian-Human Immunodeficiency Virus Infection. J. Virol. 2015, 89, 8334–8345. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Klein, F.; Lorenzi, J.C.; Seaman, M.S.; West, A.P., Jr.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2015, 522, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Schoofs, T.; Gruell, H.; Settler, A.; Karagounis, T.; Kreider, E.F.; Murrell, B.; Pfeifer, N.; Nogueira, L.; Oliveira, T.Y.; et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017, 23, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Horwitz, J.A.; Bar-On, Y.; Kreider, E.F.; Lu, C.L.; Lorenzi, J.C.; Feldmann, A.; Braunschweig, M.; Nogueira, L.; Oliveira, T.; et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 2016, 535, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Schoofs, T.; Klein, F.; Braunschweig, M.; Kreider, E.F.; Feldmann, A.; Nogueira, L.; Oliveira, T.; Lorenzi, J.C.; Parrish, E.H.; Learn, G.H.; et al. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science 2016, 352, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ramirez, M.; Garces, F.; Escolano, A.; Skog, P.; de Taeye, S.W.; Del Moral-Sanchez, I.; McGuire, A.T.; Yasmeen, A.; Behrens, A.J.; Ozorowski, G.; et al. Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J. Exp. Med. 2017, 214, 2573–2590. [Google Scholar] [CrossRef] [PubMed]
- Jardine, J.G.; Ota, T.; Sok, D.; Pauthner, M.; Kulp, D.W.; Kalyuzhniy, O.; Skog, P.D.; Thinnes, T.C.; Bhullar, D.; Briney, B.; et al. HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 2015, 349, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Jardine, J.G.; Sok, D.; Julien, J.P.; Briney, B.; Sarkar, A.; Liang, C.H.; Scherer, E.A.; Henry Dunand, C.J.; Adachi, Y.; Diwanji, D.; et al. Minimally Mutated HIV-1 Broadly Neutralizing Antibodies to Guide Reductionist Vaccine Design. PLoS Pathog. 2016, 12, e1005815. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.L.; Gorman, J.; Doria-Rose, N.A.; Morris, L. Ontogeny-based immunogens for the induction of V2-directed HIV broadly neutralizing antibodies. Immunol. Rev. 2017, 275, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Hangartner, L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu. Rev. Immunol. 2016, 34, 635–659. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Mascola, J.R. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat. Immunol. 2015, 16, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Klein, F.; Mouquet, H.; Dosenovic, P.; Scheid, J.F.; Scharf, L.; Nussenzweig, M.C. Antibodies in HIV-1 vaccine development and therapy. Science 2013, 341, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- van Gils, M.J.; Sanders, R.W. Broadly neutralizing antibodies against HIV-1: Templates for a vaccine. Virology 2013, 435, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Wilson, I.A. The HIV-1 envelope glycoprotein structure: Nailing down a moving target. Immunol. Rev. 2017, 275, 21–32. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.E.; Burton, D.R. Identification and specificity of broadly neutralizing antibodies against HIV. Immunol. Rev. 2017, 275, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Stanfield, R.L.; Wilson, I.A. Antibody vs. HIV in a clash of evolutionary titans. Proc. Natl. Acad. Sci. USA 2005, 102, 14943–14948. [Google Scholar] [CrossRef] [PubMed]
- Escolano, A.; Dosenovic, P.; Nussenzweig, M.C. Progress toward active or passive HIV-1 vaccination. J. Exp. Med. 2017, 214, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Stamatatos, L.; Morris, L.; Burton, D.R.; Mascola, J.R. Neutralizing antibodies generated during natural HIV-1 infection: Good news for an HIV-1 vaccine? Nat. Med. 2009, 15, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Van Regenmortel, M.H. Structure-Based Reverse Vaccinology Failed in the Case of HIV Because it Disregarded Accepted Immunological Theory. Int J. Mol. Sci 2016, 17, 1591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhu, J.; Yang, Y.; Gorman, J.; Ofek, G.; Srivatsan, S.; Druz, A.; Lees, C.R.; Lu, G.; Soto, C.; et al. Transplanting supersites of HIV-1 vulnerability. PLoS ONE 2014, 9, e99881. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.E.; Barouch, D.H. Broadly Neutralizing Antibodies for HIV Eradication. Curr. HIV/AIDS Rep. 2016, 13, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J. Peptides as protein binding site mimetics. Curr. Opin. Chem. Biol. 2008, 12, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.; Hirsch, T.; Overwin, H.; Eichler, J. Synthetic mimetics of the CD4 binding site of HIV-1 gp120 for the design of immunogens. Angew. Chem. Int. Ed. Engl. 2007, 46, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Rodel, K.; Kneidl, B.; Donhauser, N.; Mossl, M.; Lump, E.; Munch, J.; Schmidt, B.; Eichler, J. Enhancement and induction of HIV-1 infection through an assembled peptide derived from the CD4 binding site of gp120. Chembiochem 2015, 16, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Zwick, M.B.; Bonnycastle, L.L.; Menendez, A.; Irving, M.B.; Barbas, C.F., 3rd; Parren, P.W.; Burton, D.R.; Scott, J.K. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J. Virol. 2001, 75, 6692–6699. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Montero, M.; Menendez, A.; van Houten, N.E.; Irving, M.B.; Pantophlet, R.; Zwick, M.B.; Parren, P.W.; Burton, D.R.; Scott, J.K.; et al. Structure of a high-affinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J. Mol. Biol. 2007, 369, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Joyce, J.G.; Miller, M.D.; Finnefrock, A.C.; Liang, X.; Finotto, M.; Ingallinella, P.; McKenna, P.; Citron, M.; Ottinger, E.; et al. Vaccination with peptide mimetics of the gp41 prehairpin fusion intermediate yields neutralizing antisera against HIV-1 isolates. Proc. Natl. Acad. Sci. USA 2010, 107, 10655–10660. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.; Araujo, A.; Apellaniz, B.; Bryson, S.; Carravilla, P.; de la Arada, I.; Huarte, N.; Rujas, E.; Pai, E.F.; Arrondo, J.L.; et al. Structure and immunogenicity of a peptide vaccine, including the complete HIV-1 gp41 2F5 epitope: Implications for antibody recognition mechanism and immunogen design. J. Biol. Chem. 2014, 289, 6565–6580. [Google Scholar] [CrossRef] [PubMed]
- Poljak, R.J.; Amzel, L.M.; Chen, B.L.; Phizackerley, R.P.; Saul, F. Structure and specificity of antibody molecules. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1975, 272, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Haussner, C.; Lach, J.; Eichler, J. Synthetic antibody mimics for the inhibition of protein-ligand interactions. Curr. Opin. Chem. Biol. 2017, 40, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Haussner, C.; Damm, D.; Nirschl, S.; Rohrhofer, A.; Schmidt, B.; Eichler, J. Peptide Paratope Mimics of the Broadly Neutralizing HIV-1 Antibody b12. Chembiochem 2017, 18, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Pyati, J.; Koduri, R.; Sharp, S.J.; Thornton, G.B.; Parren, P.W.; Sawyer, L.S.; Hendry, R.M.; Dunlop, N.; Nara, P.L.; et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 1994, 266, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xu, L.; Dey, B.; Hessell, A.J.; Van Ryk, D.; Xiang, S.H.; Yang, X.; Zhang, M.Y.; Zwick, M.B.; Arthos, J.; et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 2007, 445, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Delhalle, S.; Schmit, J.C.; Chevigne, A. Phages and HIV-1: From display to interplay. Int. J. Mol. Sci. 2012, 13, 4727–4794. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Antoni, S.; Brill, B.; Landersz, M.; Rodes, B.; Soriano, V.; Wintergerst, U.; Knechten, H.; Staszewski, S.; von Laer, D.; et al. Mimotopes selected with antibodies from HIV-1-neutralizing long-term non-progressor plasma. Eur. J. Immunol. 2007, 37, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Scala, G.; Chen, X.; Liu, W.; Telles, J.N.; Cohen, O.J.; Vaccarezza, M.; Igarashi, T.; Fauci, A.S. Selection of HIV-specific immunogenic epitopes by screening random peptide libraries with HIV-1-positive sera. J. Immunol. 1999, 162, 6155–6161. [Google Scholar] [PubMed]
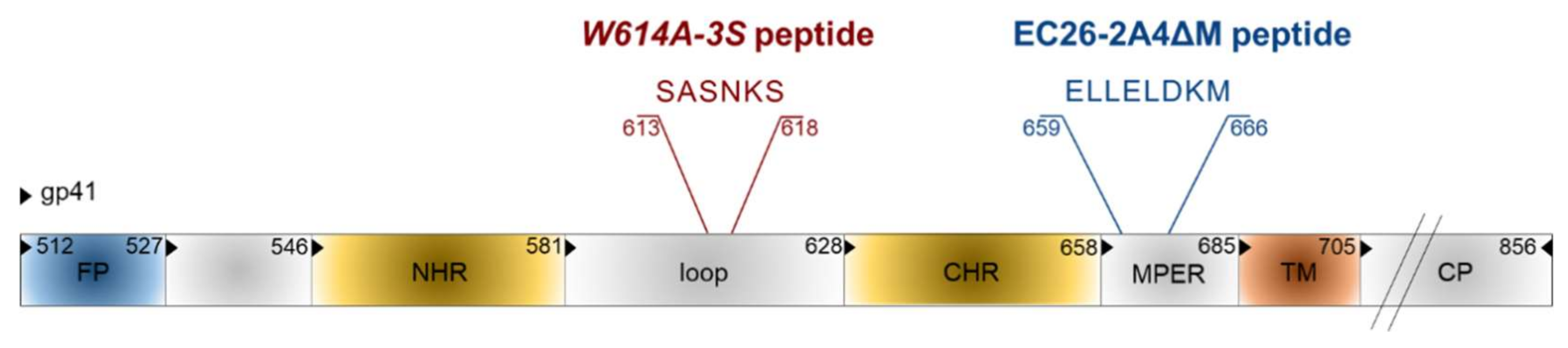
- Zhou, M.; Meyer, T.; Koch, S.; Koch, J.; von Briesen, H.; Benito, J.M.; Soriano, V.; Haberl, A.; Bickel, M.; Dubel, S.; et al. Identification of a new epitope for HIV-neutralizing antibodies in the gp41 membrane proximal external region by an Env-tailored phage display library. Eur. J. Immunol. 2013, 43, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Humbert, M.; Benz, A.; Dietrich, U. 3D-Epitope-Explorer (3DEX): Localization of conformational epitopes within three-dimensional structures of proteins. J. Comput. Chem. 2005, 26, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Kostoula, I.; Brill, B.; Panou, E.; Sakarellos-Daitsiotis, M.; Dietrich, U. Prime boost vaccination approaches with different conjugates of a new HIV-1 gp41 epitope encompassing the membrane proximal external region induce neutralizing antibodies in mice. Vaccine 2012, 30, 1911–1916. [Google Scholar] [CrossRef] [PubMed]
- Muster, T.; Steindl, F.; Purtscher, M.; Trkola, A.; Klima, A.; Himmler, G.; Ruker, F.; Katinger, H. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 1993, 67, 6642–6647. [Google Scholar] [PubMed]
- Cerutti, N.; Loredo-Varela, J.L.; Caillat, C.; Weissenhorn, W. Antigp41 membrane proximal external region antibodies and the art of using the membrane for neutralization. Curr. Opin. HIV AIDS 2017, 12, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, G.; Wiehe, K.; Nicely, N.I.; Vandergrift, N.A.; Rountree, W.; Bonsignori, M.; Alam, S.M.; Gao, J.; Haynes, B.F.; et al. Polyreactivity and autoreactivity among HIV-1 antibodies. J. Virol. 2015, 89, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Ringel, O.; Muller, K.; Koch, J.; Brill, B.; Wolf, T.; Stephan, C.; Vieillard, V.; Debre, P.; Dietrich, U. Optimization of the EC26-2A4 epitope in the gp41 membrane proximal external region (MPER) targeted by neutralizing antibodies from an elite controller. AIDS Res. Hum. Retrovir. 2018, 34, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Verkoczy, L.; Chen, Y.; Bouton-Verville, H.; Zhang, J.; Diaz, M.; Hutchinson, J.; Ouyang, Y.B.; Alam, S.M.; Holl, T.M.; Hwang, K.K.; et al. Rescue of HIV-1 broad neutralizing antibody-expressing B cells in 2F5 VH x VL knockin mice reveals multiple tolerance controls. J. Immunol. 2011, 187, 3785–3797. [Google Scholar] [CrossRef] [PubMed]
- 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm. Rep. 1992, 41, 1–19.
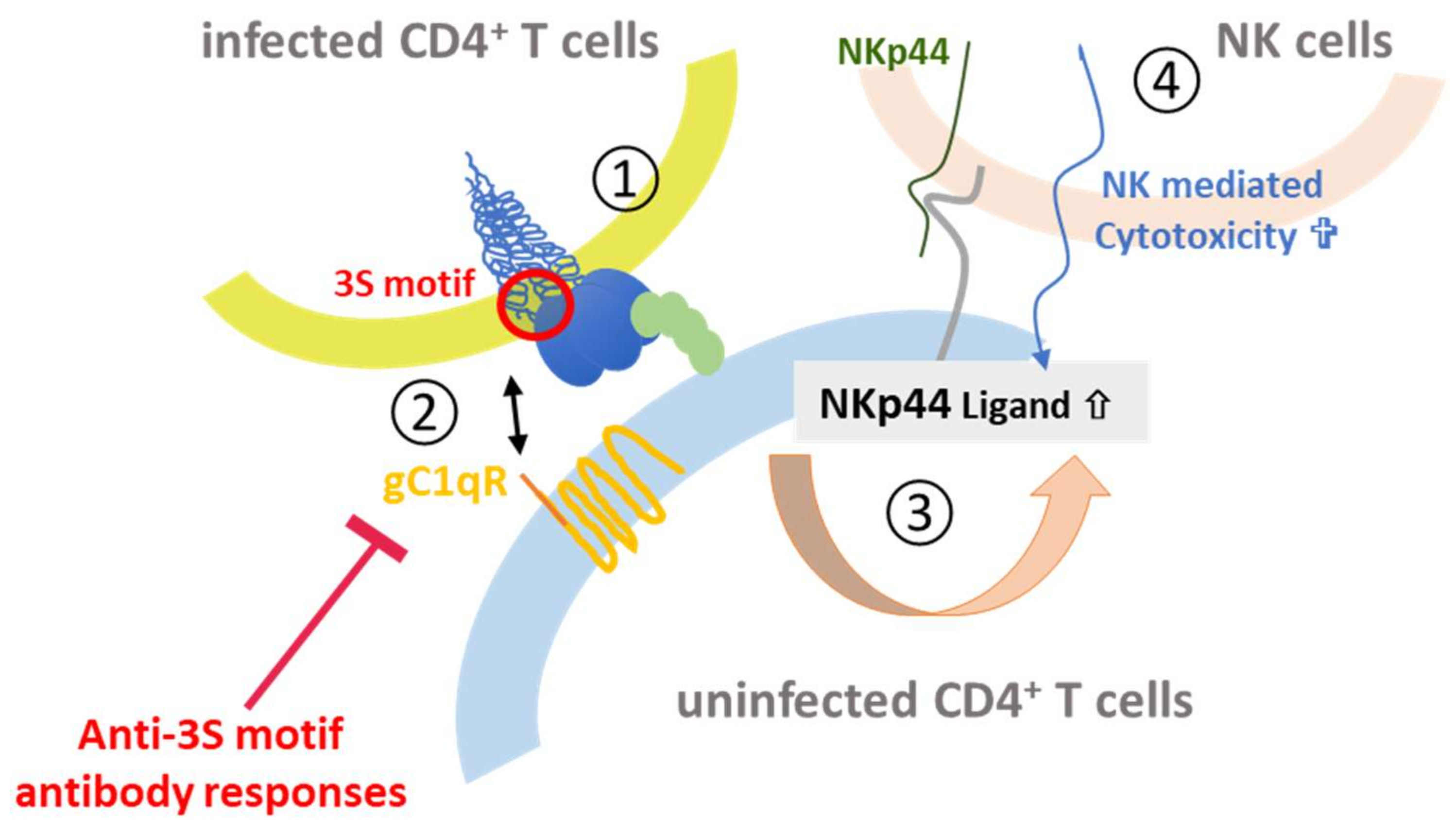
- Baychelier, F.; Sennepin, A.; Ermonval, M.; Dorgham, K.; Debre, P.; Vieillard, V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 2013, 122, 2935–2942. [Google Scholar] [CrossRef] [PubMed]
- Fausther-Bovendo, H.; Vieillard, V.; Sagan, S.; Bismuth, G.; Debre, P. HIV gp41 engages gC1qR on CD4+ T cells to induce the expression of an NK ligand through the PIP3/H2O2 pathway. PLoS Pathog. 2010, 6, e1000975. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Strominger, J.L.; Debre, P. NK cytotoxicity against CD4+ T cells during HIV-1 infection: A gp41 peptide induces the expression of an NKp44 ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 10981–10986. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Costagliola, D.; Simon, A.; Debré, P.; The French Asymtomatiques a Long Terme (ALT) Study Group. Specific adaptive humoral response against a gp41 motif inhibits CD4+ T sensitivity to NK lysis during HIV-1 infection. AIDS 2006, 20, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Crouzet, J.; Boufassa, F.; Sennepin, A.; Ho Tsong Fang, R.; Debré, P.; Meyer, L. Specific anti-gp41 antibodies predict HIV-1 disease progression. J. Acquir. Immune Defic. Syndr. 2012, 61, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Dereuddre-Bosquet, N.; Mangeot-Mederle, I.; Le Grand, R.; Debré, P. An HIV gp41 vaccine protects CD4 central memory T cells in SHIV-infected macaques. Vaccine 2012, 30, 6883–6891. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Le Grand, R.; Dausset, J.; Debré, P. A vaccine strategy against AIDS: An HIV gp41 peptide immunization prevents NKp44L expression and CD4+ T cell depletion in SHIV-infected macaques. Proc. Natl. Acad. Sci. USA 2008, 105, 2100–2104. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Gharakhanian, S.; Lucar, O.; Katlama, C.; Launay, O.; Autran, B.; Ho Tsong Fang, R.; Crouzet, J.; Murphy, R.L.; Debré, P. Perspectives for immunotherapy: Which applications might achieve an HIV functional cure? Oncotarget 2016, 7, 38946–38958. [Google Scholar] [CrossRef] [PubMed]
- Petitdemange, C.; Achour, A.; Dispinseri, S.; Malet, I.; Sennepin, A.; Ho Tsong Fang, R.; Crouzet, J.; Marcelin, A.G.; Calver, V.; Scarlatti, G.; et al. A single amino-acid change in a highly conserved motif of gp41 elicits HIV-1 neutralization and protects against CD4 depletion. Clin. Infect. Dis. 2013, 57, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Lucar, O.; Su, B.; Potard, V.; Samri, A.; Autran, B.; Moog, C.; Debré, P.; Vieillard, V. Neutralizing antibodies against a specific Human Immunodeficiency Virus gp41 epitope are associated with long-term non-progressor status. EBioMedicine 2017, 22, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, F.; Hauber, J. Antiviral therapy of persistent viral infection using genome editing. Curr. Opin. Virol. 2016, 20, 85–91. [Google Scholar] [CrossRef] [PubMed]
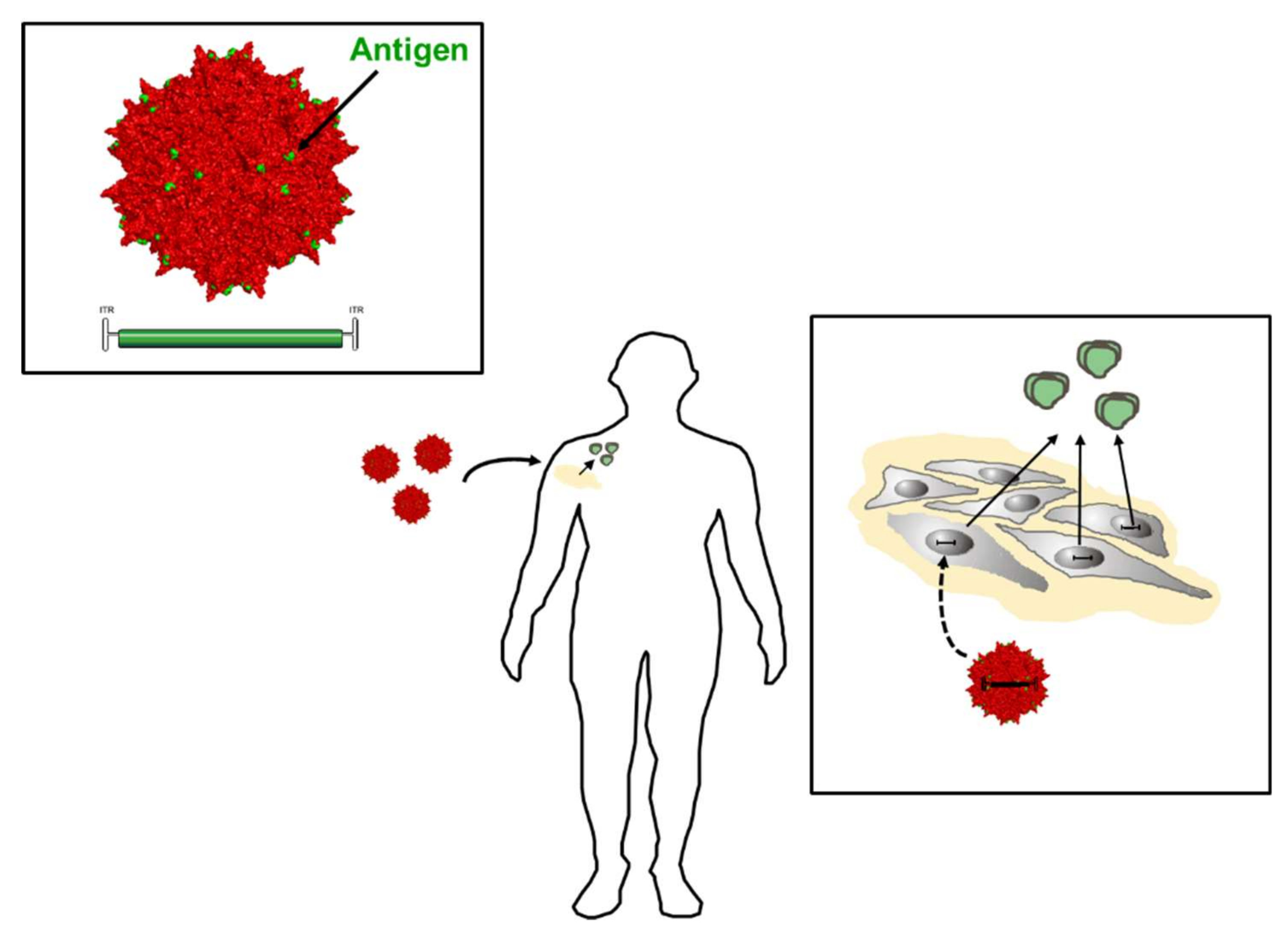
- Brady, J.M.; Baltimore, D.; Balazs, A.B. Antibody gene transfer with adeno-associated viral vectors as a method for HIV prevention. Immunol. Rev. 2017, 275, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Buning, H. Small but increasingly mighty: Latest advances in AAV vector research, design, and evolution. Hum. Gene Ther. 2017, 28, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.P.; Desrosiers, R.C. Promise and problems associated with the use of recombinant AAV for the delivery of anti-HIV antibodies. Mol. Ther. Methods Clin. Dev. 2016, 3, 16068. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Navio, J.M.; Fuchs, S.P.; Pedreno-Lopez, S.; Rakasz, E.G.; Gao, G.; Desrosiers, R.C. Host anti-antibody responses following adeno-associated virus-mediated delivery of antibodies against HIV and SIV in rhesus monkeys. Mol. Ther. 2016, 24, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Nieto, K.; Weghofer, M.; Sehr, P.; Ritter, M.; Sedlmeier, S.; Karanam, B.; Seitz, H.; Muller, M.; Kellner, M.; Horer, M.; et al. Development of AAVLP(HPV16/31L2) particles as broadly protective HPV vaccine candidate. PLoS ONE 2012, 7, e39741. [Google Scholar] [CrossRef] [PubMed]
- Rybniker, J.; Nowag, A.; Janicki, H.; Demant, K.; Hartmann, P.; Buning, H. Incorporation of antigens into viral capsids augments immunogenicity of adeno-associated virus vector-based vaccines. J. Virol. 2012, 86, 13800–13804. [Google Scholar] [CrossRef] [PubMed]
- Muhle, M.; Lehmann, M.; Hoffmann, K.; Stern, D.; Kroniger, T.; Luttmann, W.; Denner, J. Antigenic and immunosuppressive properties of a trimeric recombinant transmembrane envelope protein gp41 of HIV-1. PLoS ONE 2017, 12, e0173454. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Faingold, O.; Shai, Y. HIV-1 fusion protein exerts complex immunosuppressive effects. Trends Biochem. Sci. 2013, 38, 345–349. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ringel, O.; Vieillard, V.; Debré, P.; Eichler, J.; Büning, H.; Dietrich, U. The Hard Way towards an Antibody-Based HIV-1 Env Vaccine: Lessons from Other Viruses. Viruses 2018, 10, 197. https://doi.org/10.3390/v10040197
Ringel O, Vieillard V, Debré P, Eichler J, Büning H, Dietrich U. The Hard Way towards an Antibody-Based HIV-1 Env Vaccine: Lessons from Other Viruses. Viruses. 2018; 10(4):197. https://doi.org/10.3390/v10040197
Chicago/Turabian StyleRingel, Oliver, Vincent Vieillard, Patrice Debré, Jutta Eichler, Hildegard Büning, and Ursula Dietrich. 2018. "The Hard Way towards an Antibody-Based HIV-1 Env Vaccine: Lessons from Other Viruses" Viruses 10, no. 4: 197. https://doi.org/10.3390/v10040197
APA StyleRingel, O., Vieillard, V., Debré, P., Eichler, J., Büning, H., & Dietrich, U. (2018). The Hard Way towards an Antibody-Based HIV-1 Env Vaccine: Lessons from Other Viruses. Viruses, 10(4), 197. https://doi.org/10.3390/v10040197