The Amino-Terminal Region of Hepatitis E Virus ORF1 Containing a Methyltransferase (Met) and a Papain-Like Cysteine Protease (PCP) Domain Counteracts Type I Interferon Response

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. HEV ORF1 Cloning and Plasmid Constructs
2.3. Reagents and Antibodies
2.4. Transfections
2.5. Cell Viability Test
2.6. Immunoblot Analysis
2.7. Reporter Gene Assay
2.8. RNA Extraction, Reverse Transcription (RT) and Real-Time Quantitative PCR (RT-qPCR)
2.9. Immunostaining and Fluorescent Microscopy
2.10. Statistical Analyses
3. Results
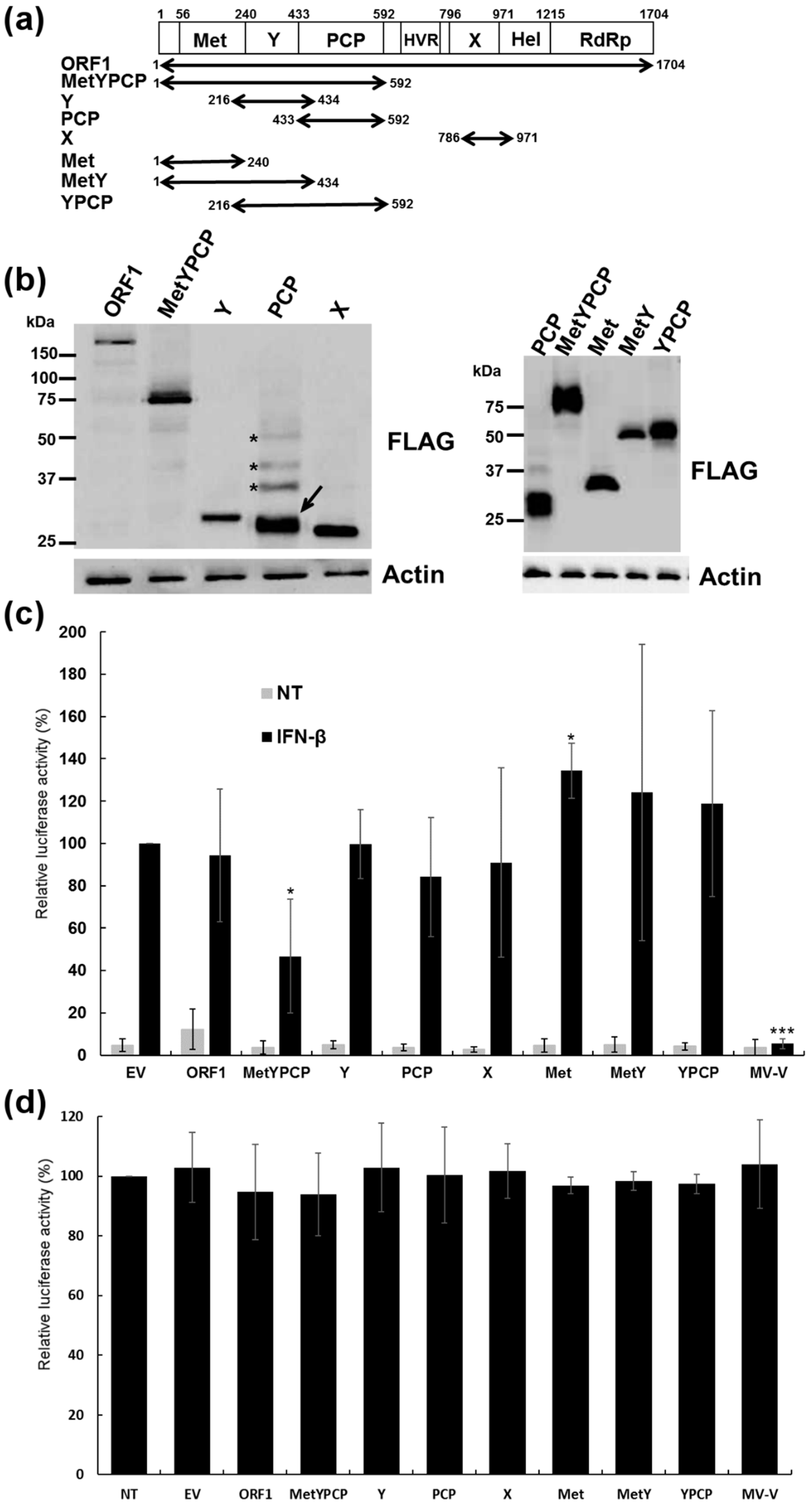
3.1. Expression of Full-Length and Individual Domains of HEV ORF1
3.2. MetYPCP of HEV ORF1 Inhibited the IFN-I Response
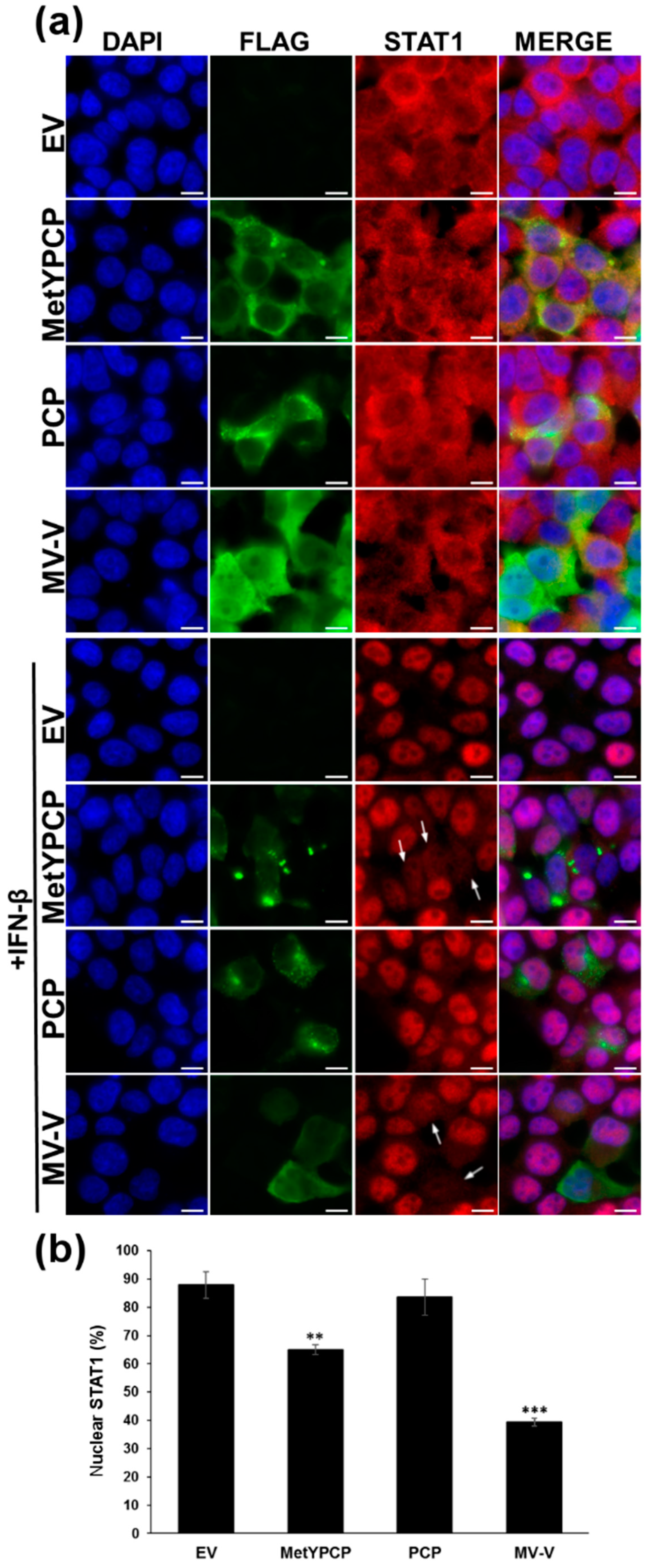
3.3. MetYPCP of HEV ORF1 Interfered with the JAK/STAT Pathway after IFN-β Treatment
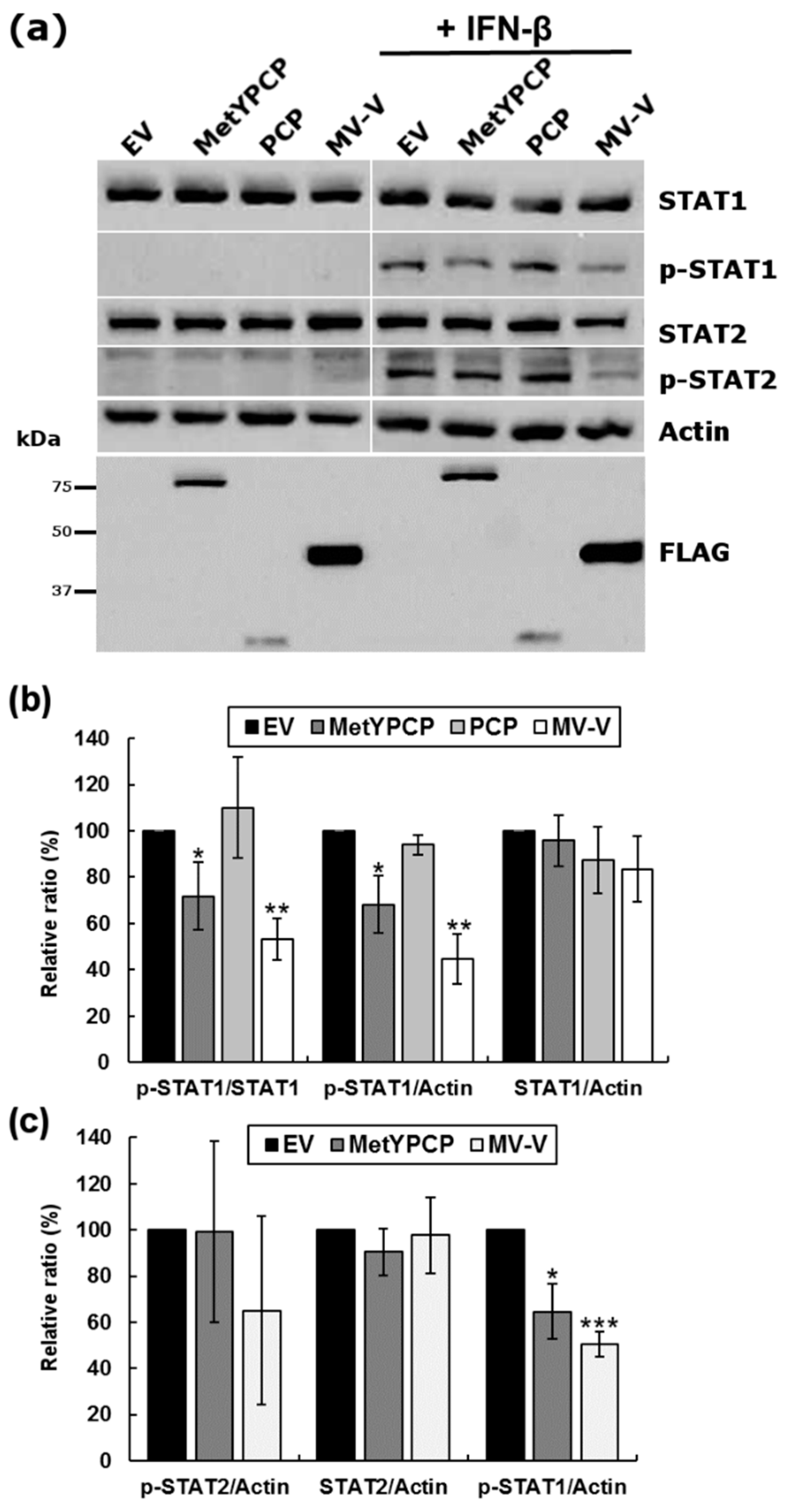
3.4. MetYPCP of HEV ORF1 Inhibited STAT1 but Not STAT2 Phosphorylation after IFN-β Treatment
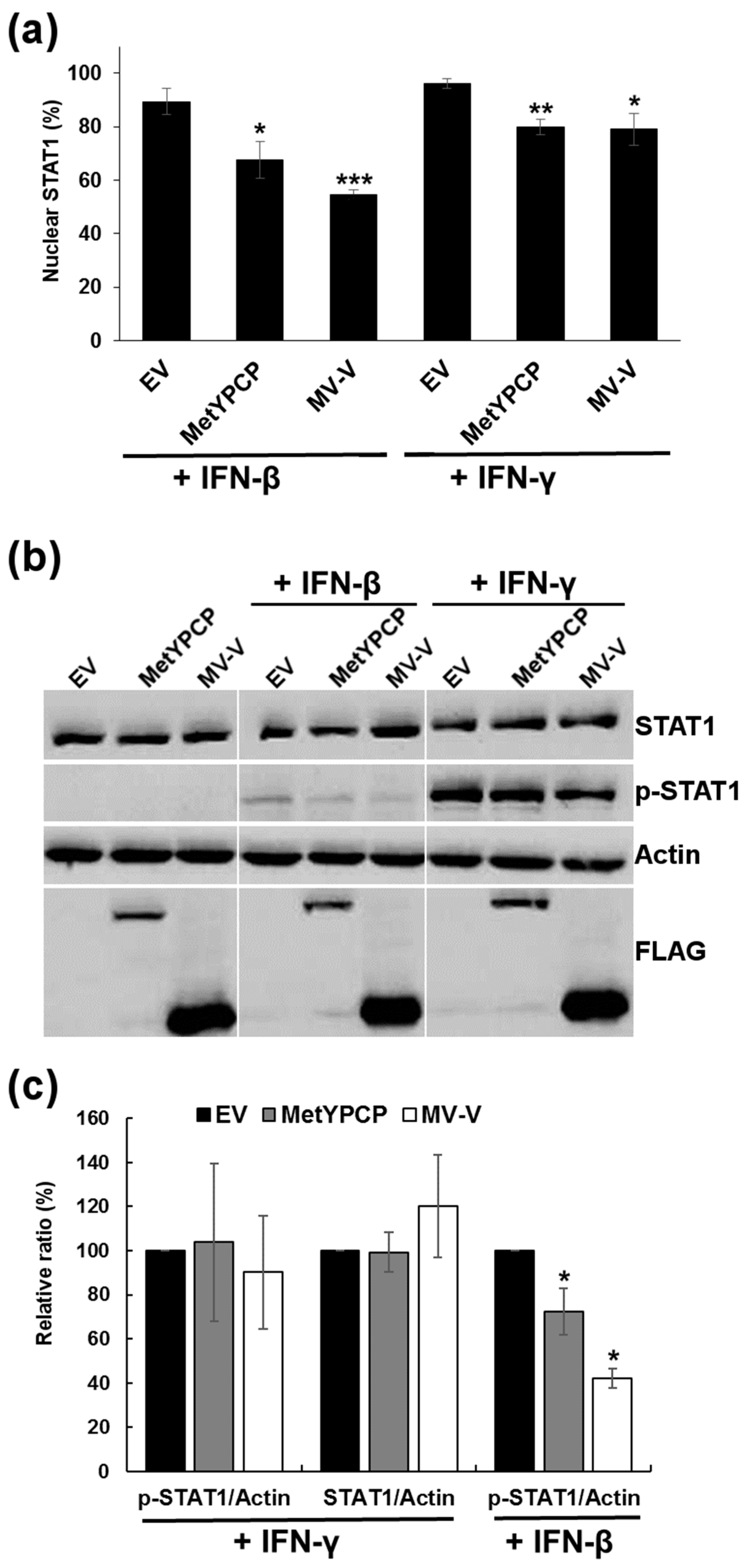
3.5. MetYPCP of HEV ORF1 Inhibited More Efficiently the JAK/STAT Pathway after IFN-I Than IFN-II Treatment
3.6. The Ability of MetYPCP of HEV ORF1 to Inhibit the JAK/STAT Pathway after IFN-I Differed between Genotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, D.B.; Simmonds, P.; International Committee on Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.-J.; Okamoto, H.; van der Poel, W.H.M.; et al. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Holla, R.P.; Jameel, S. Molecular virology of hepatitis E virus. Virus Res. 2011, 161, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Magden, J.; Takeda, N.; Li, T.; Auvinen, P.; Ahola, T.; Miyamura, T.; Merits, A.; Kääriäinen, L. Virus-specific mRNA capping enzyme encoded by hepatitis E virus. J. Virol. 2001, 75, 6249–6255. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, D.; Thomas, S.; Chakraborty, M.; Jameel, S. Expression and processing of the Hepatitis E virus ORF1 nonstructural polyprotein. Virol. J. 2006, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, D.; Panda, S.K.; Kapur, N.; Varma, S.P.K.; Durgapal, H. Hepatitis E virus (HEV) protease: A chymotrypsin-like enzyme that processes both non-structural (pORF1) and capsid (pORF2) protein. J. Gen. Virol. 2014, 95, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K. Molecular characterization of hepatitis E virus ORF1 gene supports a papain-like cysteine protease (PCP)-domain activity. Virus Res. 2013, 178, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Ropp, S.L.; Tam, A.W.; Beames, B.; Purdy, M.; Frey, T.K. Expression of the hepatitis E virus ORF1. Arch. Virol. 2000, 145, 1321–1337. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, S.; Zhou, Y.; Frey, T.K. Lack of processing of the expressed ORF1 gene product of hepatitis E virus. Virol. J. 2011, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Perttilä, J.; Spuul, P.; Ahola, T. Early secretory pathway localization and lack of processing for hepatitis E virus replication protein pORF1. J. Gen. Virol. 2013, 94, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Ansari, I.H.; Nanda, S.K.; Durgapal, H.; Agrawal, S.; Mohanty, S.K.; Gupta, D.; Jameel, S.; Panda, S.K. Cloning, sequencing, and expression of the hepatitis E virus (HEV) nonstructural open reading frame 1 (ORF1). J. Med. Virol. 2000, 60, 275–283. [Google Scholar] [CrossRef]
- Kanade, G.D.; Pingale, K.D.; Karpe, Y.A. Activities of Thrombin and Factor Xa are essential for replication of Hepatitis E virus and are possibly implicated in the ORF1 polyprotein processing. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Doceul, V.; Bagdassarian, E.; Demange, A.; Pavio, N. Zoonotic Hepatitis E Virus: Classification, Animal Reservoirs and Transmission Routes. Viruses 2016, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Pavio, N.; Meng, X.-J.; Doceul, V. Zoonotic origin of hepatitis E. Curr. Opin. Virol. 2015, 10, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Abravanel, F.; Lhomme, S.; Rostaing, L.; Izopet, J. Hepatitis E virus: Chronic infection, extra-hepatic manifestations, and treatment. Clin. Res. Hepatol. Gastroenterol. 2014, 39, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Silvennoinen, O.; Ihle, J.N.; Schlessinger, J.; Levy, D.E. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature 1993, 366, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Gauzzi, M.C.; Velazquez, L.; McKendry, R.; Mogensen, K.E.; Fellous, M.; Pellegrini, S. Interferon-α-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J. Biol. Chem. 1996, 271, 20494–20500. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.; Yan, H.; Lim, J.T.; Krolewski, J.J. Dimerization of a chimeric CD4-interferon-α receptor reconstitutes the signaling events preceding STAT phosphorylation. Oncogene 1996, 13, 125–133. [Google Scholar] [PubMed]
- Platanias, L.C.; Uddin, S.; Colamonici, O.R. Tyrosine phosphorylation of the α and β subunits of the type I interferon receptor. Interferon-β selectively induces tyrosine phosphorylation of an α subunit-associated protein. J. Biol. Chem. 1994, 269, 17761–17764. [Google Scholar] [PubMed]
- Zhao, W.; Lee, C.; Piganis, R.; Plumlee, C.; de Weerd, N.; Hertzog, P.J.; Schindler, C. A conserved IFN-α receptor tyrosine motif directs the biological response to type I IFNs. J. Immunol. 2008, 180, 5483–5489. [Google Scholar] [CrossRef] [PubMed]
- Abramovich, C.; Shulman, L.M.; Ratovitski, E.; Harroch, S.; Tovey, M.; Eid, P.; Revel, M. Differential tyrosine phosphorylation of the IFNAR chain of the type I interferon receptor and of an associated surface protein in response to IFN-α and IFN-β. EMBO J. 1994, 13, 5871–5877. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C.; Colamonici, O.R. Interferon α induces rapid tyrosine phosphorylation of the α subunit of its receptor. J. Biol. Chem. 1992, 267, 24053–24057. [Google Scholar] [PubMed]
- Yan, H.; Krishnan, K.; Greenlund, A.C.; Gupta, S.; Lim, J.T.; Schreiber, R.D.; Schindler, C.W.; Krolewski, J.J. Phosphorylated interferon-α receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 1996, 15, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Najjar, I.; Fagard, R. STAT1 and pathogens, not a friendly relationship. Biochimie 2010, 92, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Ha, Y.; Chae, C. Expression of interferon-α and Mx protein in the livers of pigs experimentally infected with swine hepatitis E virus. J. Comp. Pathol. 2010, 142, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Moal, V.; Textoris, J.; Ben Amara, A.; Mehraj, V.; Berland, Y.; Colson, P.; Mege, J.-L. Chronic hepatitis E virus infection is specifically associated with an interferon-related transcriptional program. J. Infect. Dis. 2013, 207, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qi, Y.; Harrison, T.J.; Luo, B.; Zhou, Y.; Li, X.; Song, A.; Huang, W.; Wang, Y. Hepatitis E genotype 4 virus from feces of monkeys infected experimentally can be cultured in PLC/PRF/5 cells and upregulate host interferon-inducible genes. J. Med. Virol. 2014, 86, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.W.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; François, C.; Anggakusuma; Behrendt, P.; Engelmann, M.; Knegendorf, L.; Vieyres, G.; Wedemeyer, H.; Hartmann, R.; Pietschmann, T.; et al. Antiviral Activities of Different Interferon Types and Subtypes against Hepatitis E Virus Replication. Antimicrob. Agents Chemother. 2016, 60, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zafrullah, M.; Mixson-Hayden, T.; Dai, X.; Liang, J.; Meng, J.; Kamili, S. Suppression of interferon-α signaling by hepatitis E virus. Hepatology 2012, 55, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.-J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Choi, C.; Choi, I.; Han, K.-N.; Roh, S.W.; Choi, J.; Kwon, J.; Park, M.-K.; Kim, S.-J.; Myoung, J. Hepatitis E Virus Methyltransferase Inhibits Type I Interferon Induction by Targeting RIG-I. J. Microbiol. Biotechnol. 2018, 28, 1554–1562. [Google Scholar] [PubMed]
- Lenggenhager, D.; Gouttenoire, J.; Malehmir, M.; Bawohl, M.; Honcharova-Biletska, H.; Kreutzer, S.; Semela, D.; Neuweiler, J.; Hürlimann, S.; Aepli, P.; et al. Visualization of hepatitis E virus RNA and proteins in the human liver. J. Hepatol. 2017, 67, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. MBio 2016, 7, e01721-16. [Google Scholar] [CrossRef] [PubMed]
- Kuri, T.; Eriksson, K.K.; Putics, A.; Züst, R.; Snijder, E.J.; Davidson, A.D.; Siddell, S.G.; Thiel, V.; Ziebuhr, J.; Weber, F. The ADP-ribose-1″-monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J. Gen. Virol. 2011, 92, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Griffin, D.E. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, Y.; Ransburgh, R.; Snijder, E.J.; Fang, Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J. Virol. 2012, 86, 3839–3850. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, G.; Zheng, D.; Cheng, G.; Tang, H. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS ONE 2011, 6, e17192. [Google Scholar] [CrossRef] [PubMed]
- Karpe, Y.A.; Lole, K.S. Deubiquitination activity associated with hepatitis E virus putative papain-like cysteine protease. J. Gen. Virol. 2011, 92, 2088–2092. [Google Scholar] [CrossRef] [PubMed]
- Caignard, G.; Guerbois, M.; Labernardière, J.-L.; Jacob, Y.; Jones, L.M.; Infectious Mapping Project I-MAP; Wild, F.; Tangy, F.; Vidalain, P.-O. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-α/β signaling. Virology 2007, 368, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.-A.; Jacob, Y.; Cassonnet, P.; Favre, M. Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor β signaling pathway by binding to SMAD3. J. Virol. 2006, 80, 12420–12424. [Google Scholar] [CrossRef] [PubMed]
- Doceul, V.; Chauveau, E.; Lara, E.; Breard, E.; Sailleau, C.; Zientara, S.; Vitour, D. Dual modulation of type I interferon response by Bluetongue virus. J. Virol. 2014, 88, 10792–10802. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bagdassarian, E. Interaction of Hepatitis E Virus with the Host Interferon Response. Ph.D. Thesis, Paris-Saclay University, Paris, France, 2017. [Google Scholar]
- Marg, A.; Shan, Y.; Meyer, T.; Meissner, T.; Brandenburg, M.; Vinkemeier, U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J. Cell Biol. 2004, 165, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Kadota, S.; Takeda, M.; Miyajima, N.; Nagata, K. Measles virus V protein blocks interferon (IFN)-α/β but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003, 545, 177–182. [Google Scholar] [CrossRef]
- Chinnakannan, S.K.; Nanda, S.K.; Baron, M.D. Morbillivirus v proteins exhibit multiple mechanisms to block type 1 and type 2 interferon signalling pathways. PLoS ONE 2013, 8, e57063. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Parisien, J.-P.; Horvath, C.M. STAT2 is a primary target for measles virus V protein-mediated α/β interferon signaling inhibition. J. Virol. 2008, 82, 8330–8338. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.-H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Soomro, M.H.; Shi, R.; She, R.; Yang, Y.; Hu, F.; Li, H. Antigen detection and apoptosis in Mongolian gerbil’s kidney experimentally intraperitoneally infected by swine hepatitis E virus. Virus Res. 2016, 213, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Zhao, C.; Huang, W.; Harrison, T.J.; Zhang, H.; Geng, K.; Wang, Y. Detection and assessment of infectivity of hepatitis E virus in urine. J. Hepatol. 2016, 64, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.P.; Kasorndorkbua, C.; Halbur, P.G.; Haqshenas, G.; Guenette, D.K.; Toth, T.E.; Meng, X.J. Evidence of extrahepatic sites of replication of the hepatitis E virus in a swine model. J. Clin. Microbiol. 2001, 39, 3040–3046. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lei, Y.; Liu, L.; Liu, P.; Xia, J.; Zhang, Y.; Zeng, H.; Wang, L.; Wang, L.; Zhuang, H. SPF rabbits infected with rabbit hepatitis E virus isolate experimentally showing the chronicity of hepatitis. PLoS ONE 2014, 9, e99861. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, J.; Wang, L.; Wang, Y. Experimental infection of rabbits with genotype 3 hepatitis E virus produced both chronicity and kidney injury. Gut 2017, 66, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; García-Sastre, A. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 2010, 13, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, X.; Wang, F.; Wang, L.; Qiao, S.; Guo, J.; Luo, C.; Wan, B.; Deng, R.; Zhang, G. The zinc-finger domain was essential for porcine reproductive and respiratory syndrome virus nonstructural protein-1α to inhibit the production of interferon-β. J. Interf. Cytokine Res. 2013, 33, 328–334. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Product | Primers |
|---|---|
| ORF1 | F: GGGGACAACTTTGTACAAAAAAGTTGGCATGGAGGCCCACCAGTTCATT R: GGGGACAACTTTGTACAAGAAAGTTGGTCATTCCAACCTCTGTATGAT |
| Met | F: GGGGACAACTTTGTACAAAAAAGTTGGCATGGAGGCCCACCAGTTCATT R: GGGGACAACTTTGTACAAGAAAGTTGGTTAGATCCATGCACGAAGTATAG |
| Y | F: GGGGACAACTTTGTACAAAAAAGTTGGCCGCGCCGTCGTGACTTATGAG R: GGGGACAACTTTGTACAAGAAAGTTGGTTAGCACTGTGCATAAAACTGTAG |
| PCP | F: GGGGACAACTTTGTACAAAAAAGTTGGCCAGTGCCGCCGCTGGCTCTCA R: GGGGACAACTTTGTACAAGAAAGTTGGTTACAAAACATACTGTTCGGGACCGTTG |
| MetYPCP | F: GGGGACAACTTTGTACAAAAAAGTTGGCATGGAGGCCCACCAGTTCATT R: GGGGACAACTTTGTACAAGAAAGTTGGTTACAAAACATACTGTTCGGGACCGTTG |
| X | F: GGGGACAACTTTGTACAAAAAAGTTGGCGCCCGCACTCGCCGGCTCCTT R: GGGGACAACTTTGTACAAGAAAGTTGGTTAGCCGGCGCAAGCACGACCCAC |
| MetY | F: GGGGACAACTTTGTACAAAAAAGTTGGCATGGAGGCCCACCAGTTCATT R: GGGGACAACTTTGTACAAGAAAGTTGGTTAGCACTGTGCATAAAACTGTAG |
| YPCP | F: GGGGACAACTTTGTACAAAAAAGTTGGCCGCGCCGTCGTGACTTATGAG R: GGGGACAACTTTGTACAAGAAAGTTGGTTACAAAACATACTGTTCGGGACCGTTG |
| MetYPCP (HEV-1) | F: GGGGACAACTTTGTACAAAAAAGTTGGCATGGAGGCCCATCAGTTTATCAAG R: GGGGACAACTTTGTACAAGAAAGTTGGTTAAAGATTGTGGCGCTCCGGGC |
| PCP (HEV-1) | F: GGGGACAACTTTGTACAAAAAAGTTGGCCAGTGTAGGCGCTGGCTTTCG R: GGGGACAACTTTGTACAAGAAAGTTGGTTAAAGATTGTGGCGCTCCGGGC |
| ISG56 | F: GGACAGGAAGCTGAAGGAG R: AGTGGGTGTTTCCTGCAA |
| MDA5 | F: ACACGTTCTTTGCGATTTCC R: ACCAAATACAGGAGCCATGC |
| OAS1 | F: CATCCGCCTAGTCAAGCACTG R: CACCACCCAAGTTTCCTGTAG |
| GADPH | F: GGTCGGAGGTCAACGGATTTG R: ACTCCACGACGTACTCAGCG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagdassarian, E.; Doceul, V.; Pellerin, M.; Demange, A.; Meyer, L.; Jouvenet, N.; Pavio, N. The Amino-Terminal Region of Hepatitis E Virus ORF1 Containing a Methyltransferase (Met) and a Papain-Like Cysteine Protease (PCP) Domain Counteracts Type I Interferon Response. Viruses 2018, 10, 726. https://doi.org/10.3390/v10120726
Bagdassarian E, Doceul V, Pellerin M, Demange A, Meyer L, Jouvenet N, Pavio N. The Amino-Terminal Region of Hepatitis E Virus ORF1 Containing a Methyltransferase (Met) and a Papain-Like Cysteine Protease (PCP) Domain Counteracts Type I Interferon Response. Viruses. 2018; 10(12):726. https://doi.org/10.3390/v10120726
Chicago/Turabian StyleBagdassarian, Eugénie, Virginie Doceul, Marie Pellerin, Antonin Demange, Léa Meyer, Nolwenn Jouvenet, and Nicole Pavio. 2018. "The Amino-Terminal Region of Hepatitis E Virus ORF1 Containing a Methyltransferase (Met) and a Papain-Like Cysteine Protease (PCP) Domain Counteracts Type I Interferon Response" Viruses 10, no. 12: 726. https://doi.org/10.3390/v10120726
APA StyleBagdassarian, E., Doceul, V., Pellerin, M., Demange, A., Meyer, L., Jouvenet, N., & Pavio, N. (2018). The Amino-Terminal Region of Hepatitis E Virus ORF1 Containing a Methyltransferase (Met) and a Papain-Like Cysteine Protease (PCP) Domain Counteracts Type I Interferon Response. Viruses, 10(12), 726. https://doi.org/10.3390/v10120726