Tet-Inducible Production of Infectious Zika Virus from the Full-Length cDNA Clones of African- and Asian-Lineage Strains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. DNA Construction
2.3. DNA Mutagenesis
2.4. DNA Transfection and Virus Production
2.5. Immunofluorescence Assay (IFA)
2.6. Virus Titration and Plaque Assay
2.7. Determination of the Stability of Infectious Zika Virus (ZIKV) cDNA Clone
2.8. Statistical Analysis
3. Results
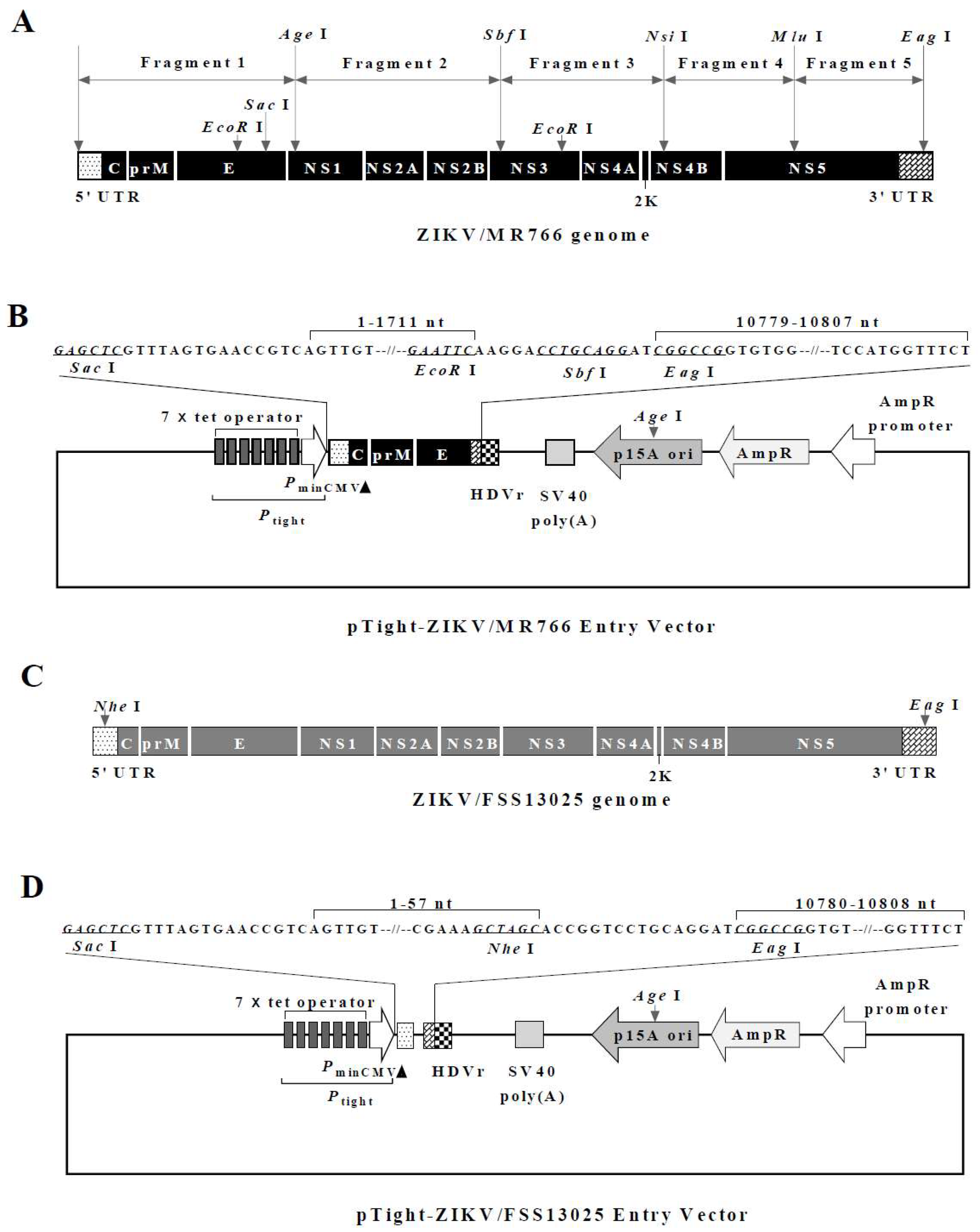
3.1. Construction of the Full-Length Infectious ZIKV cDNA
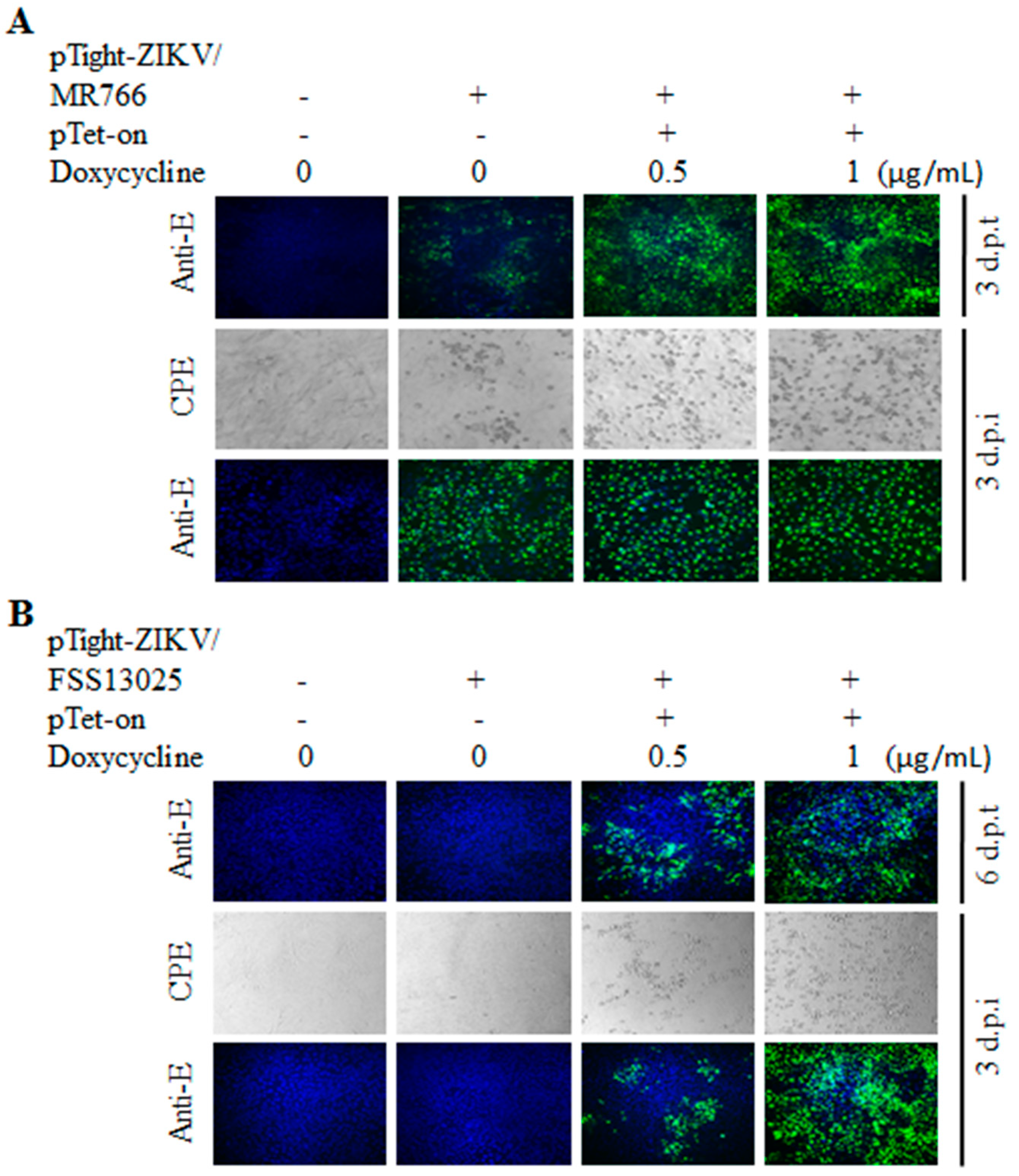
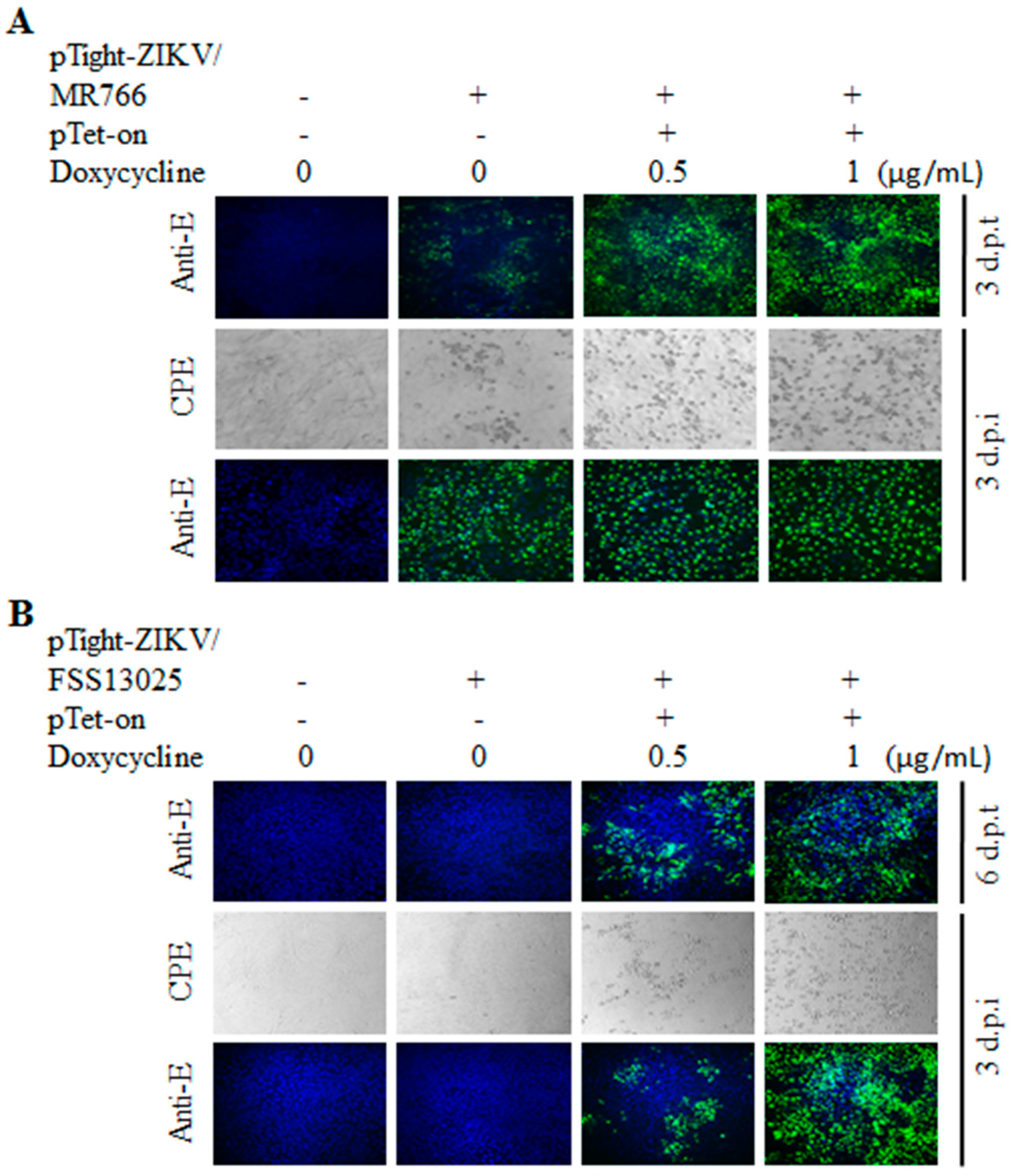
3.2. Tet-Inducible Production of Infectious ZIKV from Its cDNA
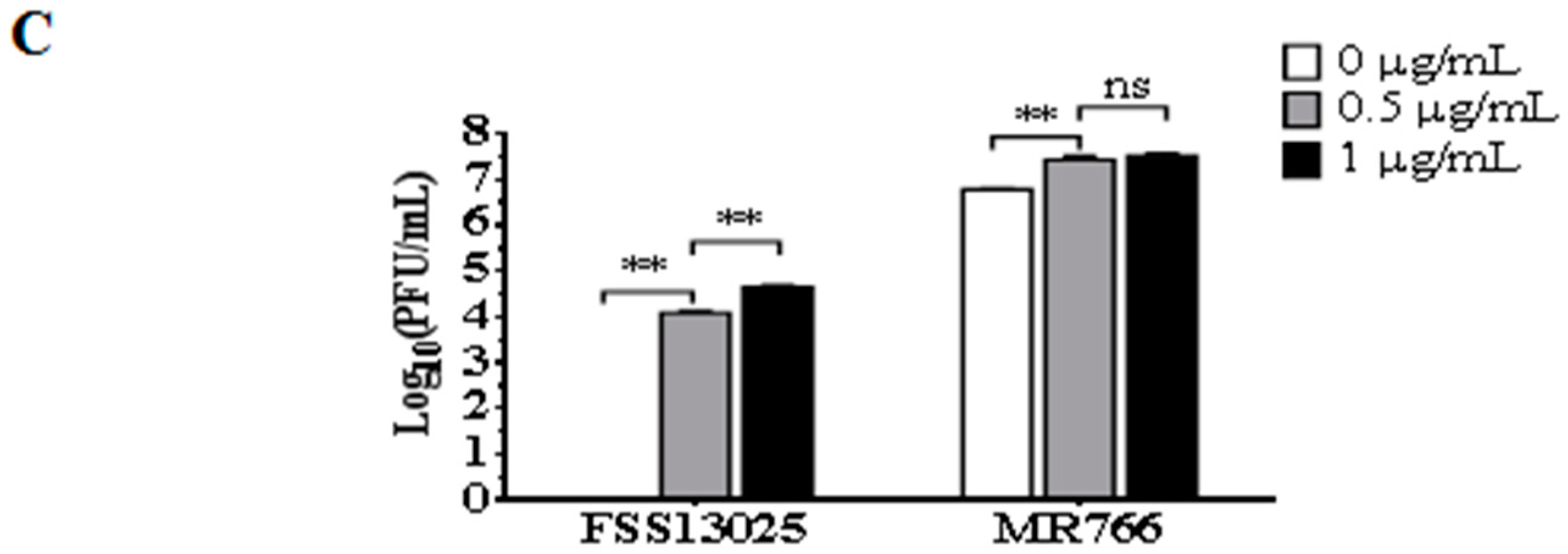
3.3. Comparison of Virus Growth between the cDNA-Derived MR766 and FSS13025 Viruses
3.4. Validation of Infectious ZIKV by the Identification of Genetic Markers
3.5. Stability of Infectious ZIKV cDNA in E. coli
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Abushouk, A.I.; Negida, A.; Ahmed, H. An updated review of Zika virus. J. Clin. Virol. 2016, 84, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martinez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N.; Group WHOZCW. Zika virus Infection as a Cause of Congenital Brain Abnormalities and Guillain-Barre Syndrome: Systematic Review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Sequeira, P.C.; Freitas, A.D.; Zogbi, H.E.; Calvet, G.A.; de Souza, R.V.; Siqueira, A.M.; de Mendonca, M.C.; Nogueira, R.M.; de Filippis, A.M.; et al. Guillain-Barre syndrome associated with Zika virus infection. Lancet 2016, 387, 1482. [Google Scholar] [CrossRef]
- Aid, M.; Abbink, P.; Larocca, R.A.; Boyd, M.; Nityanandam, R.; Nanayakkara, O.; Martinot, A.J.; Moseley, E.T.; Blass, E.; Borducchi, E.N.; et al. Zika Virus Persistence in the Central Nervous System and Lymph Nodes of Rhesus Monkeys. Cell 2017, 169, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Paz-Bailey, G.; Rosenberg, E.S.; Doyle, K.; Munoz-Jordan, J.; Santiago, G.A.; Klein, L.; Perez-Padilla, J.; Medina, F.A.; Waterman, S.H.; Gubern, C.G.; et al. Persistence of Zika Virus in Body Fluids—Preliminary Report. N. Engl. J. Med. 2018, 379, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Musso, D. Zika virus Transmission from French Polynesia to Brazil. Emerg. Infect. Dis. 2015, 21, 1887. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Young, M.P.; Mamidi, A.; Regla-Nava, J.A.; Kim, K.; Shresta, S. A Mouse Model of Zika Virus Sexual Transmission and Vaginal Viral Replication. Cell Rep. 2016, 17, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Jurado, K.A.; Hwang, J.; Szigeti-Buck, K.; Horvath, T.L.; Iwasaki, A.; Fikrig, E. Fetal Growth Restriction Caused by Sexual Transmission of Zika Virus in Mice. J. Infect. Dis. 2017, 215, 1720–1724. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Zika virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef]
- Duggal, N.K.; Ritter, J.M.; McDonald, E.M.; Romo, H.; Guirakhoo, F.; Davis, B.S.; Chang, G.J.; Brault, A.C. Differential Neurovirulence of African and Asian Genotype Zika virus Isolates in Outbred Immunocompetent Mice. Am. J. Trop. Med. Hyg. 2017, 97, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Govero, J.; Esakky, P.; Scheaffer, S.M.; Fernandez, E.; Drury, A.; Platt, D.J.; Gorman, M.J.; Richner, J.M.; Caine, E.A.; Salazar, V.; et al. Zika virus infection damages the testes in mice. Nature 2016, 540, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; van Riel, D.; Van de Perre, P.; Rockx, B.; Salinas, S. Differential virulence between Asian and African lineages of Zika virus. PLoS Negl. Trop. Dis. 2017, 11, e0005821. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere 2016, 1. [Google Scholar] [CrossRef]
- Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Luo, H.; Xie, X.; Medeiros, D.B.A.; Wakamiya, M.; Tesh, R.B.; Barrett, A.D.; Wang, T.; et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat. Med. 2017, 23, 763–767. [Google Scholar] [CrossRef]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Kenney, H.; Chen, R.; Liu, G.; Manukyan, H.; Whitehead, S.S.; Laassri, M.; Chumakov, K.; Pletnev, A.G. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 Epidemic in Brazil as a Genetic Platform for Studies of Virus-Host Interactions and Vaccine Development. mBio 2016, 7. [Google Scholar] [CrossRef]
- Weger-Lucarelli, J.; Duggal, N.K.; Bullard-Feibelman, K.; Veselinovic, M.; Romo, H.; Nguyen, C.; Ruckert, C.; Brault, A.C.; Bowen, R.A.; Stenglein, M.; et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Widman, D.G.; Young, E.; Yount, B.L.; Plante, K.S.; Gallichotte, E.N.; Carbaugh, D.L.; Peck, K.M.; Plante, J.; Swanstrom, J.; Heise, M.T.; et al. A Reverse Genetics Platform That Spans the Zika Virus Family Tree. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Aaskov, J.; Lott, W.B. Identification of a cryptic prokaryotic promoter within the cDNA encoding the 5′ end of dengue virus RNA genome. PLoS ONE 2011, 6, e18197. [Google Scholar] [CrossRef] [PubMed]
- Munster, M.; Plaszczyca, A.; Cortese, M.; Neufeldt, C.J.; Goellner, S.; Long, G.; Bartenschlager, R. A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy. Viruses 2018, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Ruggli, N.; Rice, C.M. Functional cDNA clones of the Flaviviridae: Strategies and applications. Adv. Virus Res. 1999, 53, 183–207. [Google Scholar] [PubMed]
- Liu, Z.Y.; Yu, J.Y.; Huang, X.Y.; Fan, H.; Li, X.F.; Deng, Y.Q.; Ji, X.; Cheng, M.L.; Ye, Q.; Zhao, H.; et al. Characterization of cis-Acting RNA Elements of Zika Virus by Using a Self-Splicing Ribozyme-Dependent Infectious Clone. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.L.; Zhang, Q.Y.; Chen, D.D.; Liu, S.Q.; Qin, C.F.; Zhang, B.; Ye, H.Q. Recovery of the Zika virus through an in vitro ligation approach. J. Gen. Virol. 2017, 98, 1739–1743. [Google Scholar] [CrossRef]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, C.; Chang, K.S.; Jiang, J.; Ahn, B.C.; Wakita, T.; Liang, T.J.; Luo, G. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J. Virol. 2005, 79, 13963–13973. [Google Scholar] [CrossRef]
- Anfasa, F.; Siegers, J.Y.; van der Kroeg, M.; Mumtaz, N.; Stalin Raj, V.; de Vrij, F.M.S.; Widagdo, W.; Gabriel, G.; Salinas, S.; Simonin, Y.; et al. Phenotypic Differences between Asian and African Lineage Zika Viruses in Human Neural Progenitor Cells. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Sheridan, M.A.; Balaraman, V.; Schust, D.J.; Ezashi, T.; Roberts, R.M.; Franz, A.W.E. African and Asian strains of Zika virus differ in their ability to infect and lyse primitive human placental trophoblast. PLoS ONE 2018, 13, e0200086. [Google Scholar] [CrossRef]
- Simonin, Y.; Loustalot, F.; Desmetz, C.; Foulongne, V.; Constant, O.; Fournier-Wirth, C.; Leon, F.; Moles, J.P.; Goubaud, A.; Lemaitre, J.M.; et al. Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine 2016, 12, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Cruz-Cosme, R.; Armstrong, N.; Obwolo, L.A.; Wen, F.; Hu, W.; Luo, M.H.; Tang, Q. Molecular cloning and characterization of the genes encoding the proteins of Zika virus. Gene 2017, 628, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, B.O.; Sachs, D.; Schwarz, M.C.; Palese, P.; Evans, M.J. Transposon mutagenesis of the Zika virus genome highlights regions essential for RNA replication and restricted for immune evasion. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aubry, F.; Nougairede, A.; Gould, E.A.; de Lamballerie, X. Flavivirus reverse genetic systems, construction techniques and applications: A historical perspective. Antivir. Res. 2015, 114, 67–85. [Google Scholar] [CrossRef] [Green Version]
- Lewin, A.; Mayer, M.; Chusainow, J.; Jacob, D.; Appel, B. Viral promoters can initiate expression of toxin genes introduced into Escherichia coli. BMC Biotechnol. 2005, 5, 19. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ji, W.; Lyu, S.; Qiao, L.; Luo, G. Tet-Inducible Production of Infectious Zika Virus from the Full-Length cDNA Clones of African- and Asian-Lineage Strains. Viruses 2018, 10, 700. https://doi.org/10.3390/v10120700
Zhang L, Ji W, Lyu S, Qiao L, Luo G. Tet-Inducible Production of Infectious Zika Virus from the Full-Length cDNA Clones of African- and Asian-Lineage Strains. Viruses. 2018; 10(12):700. https://doi.org/10.3390/v10120700
Chicago/Turabian StyleZhang, Lizhou, Wei Ji, Shuang Lyu, Luhua Qiao, and Guangxiang Luo. 2018. "Tet-Inducible Production of Infectious Zika Virus from the Full-Length cDNA Clones of African- and Asian-Lineage Strains" Viruses 10, no. 12: 700. https://doi.org/10.3390/v10120700
APA StyleZhang, L., Ji, W., Lyu, S., Qiao, L., & Luo, G. (2018). Tet-Inducible Production of Infectious Zika Virus from the Full-Length cDNA Clones of African- and Asian-Lineage Strains. Viruses, 10(12), 700. https://doi.org/10.3390/v10120700