Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.)


 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
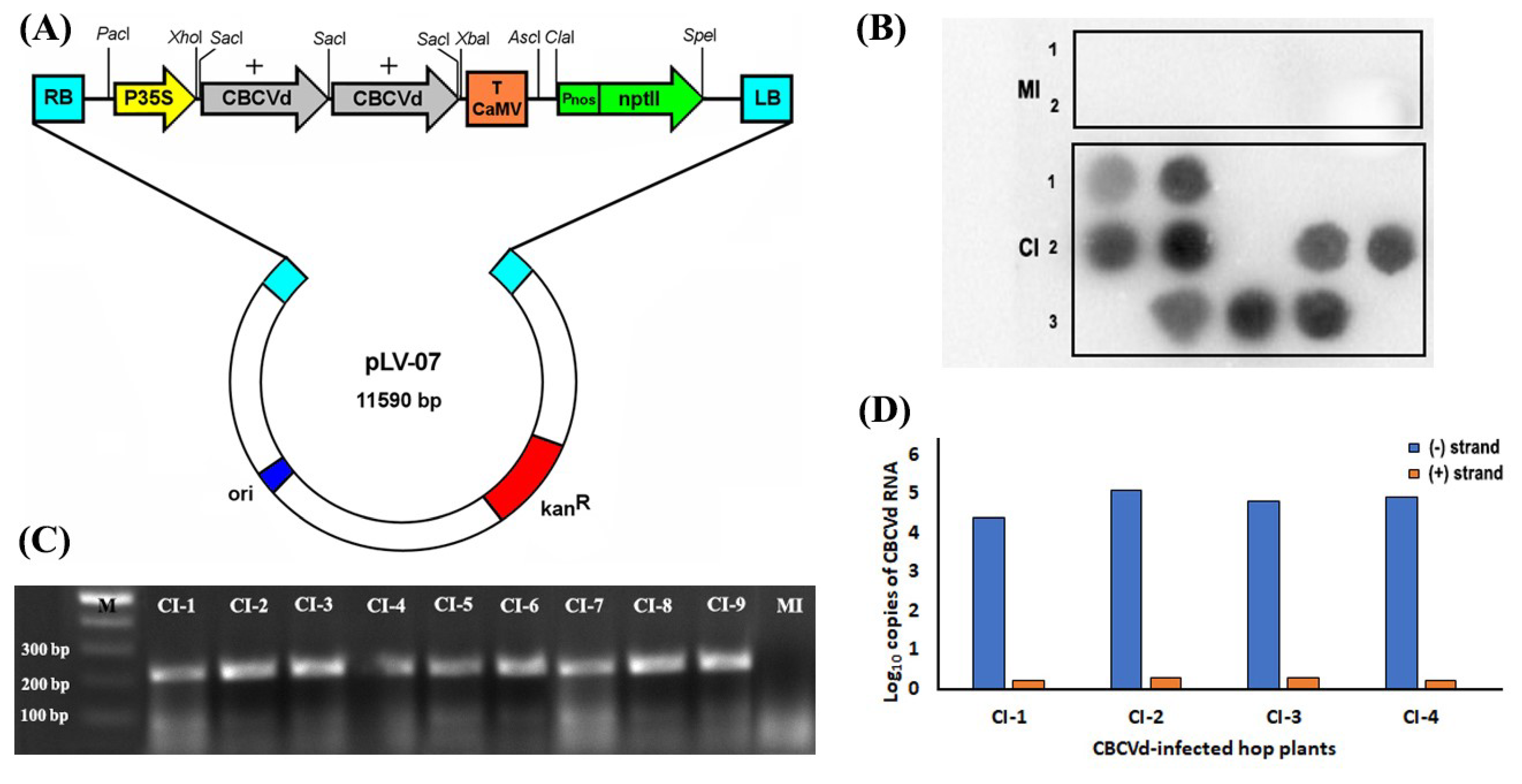
2.1. Preparation of Dimeric CBCVd Construct and Inoculation of the Hop Plants
2.2. RNA Isolation, Detection and Quantification of Genomic RNA of CBCVd
2.3. Transcriptome Sequencing, Assembly and Differential Gene Expression Profiling
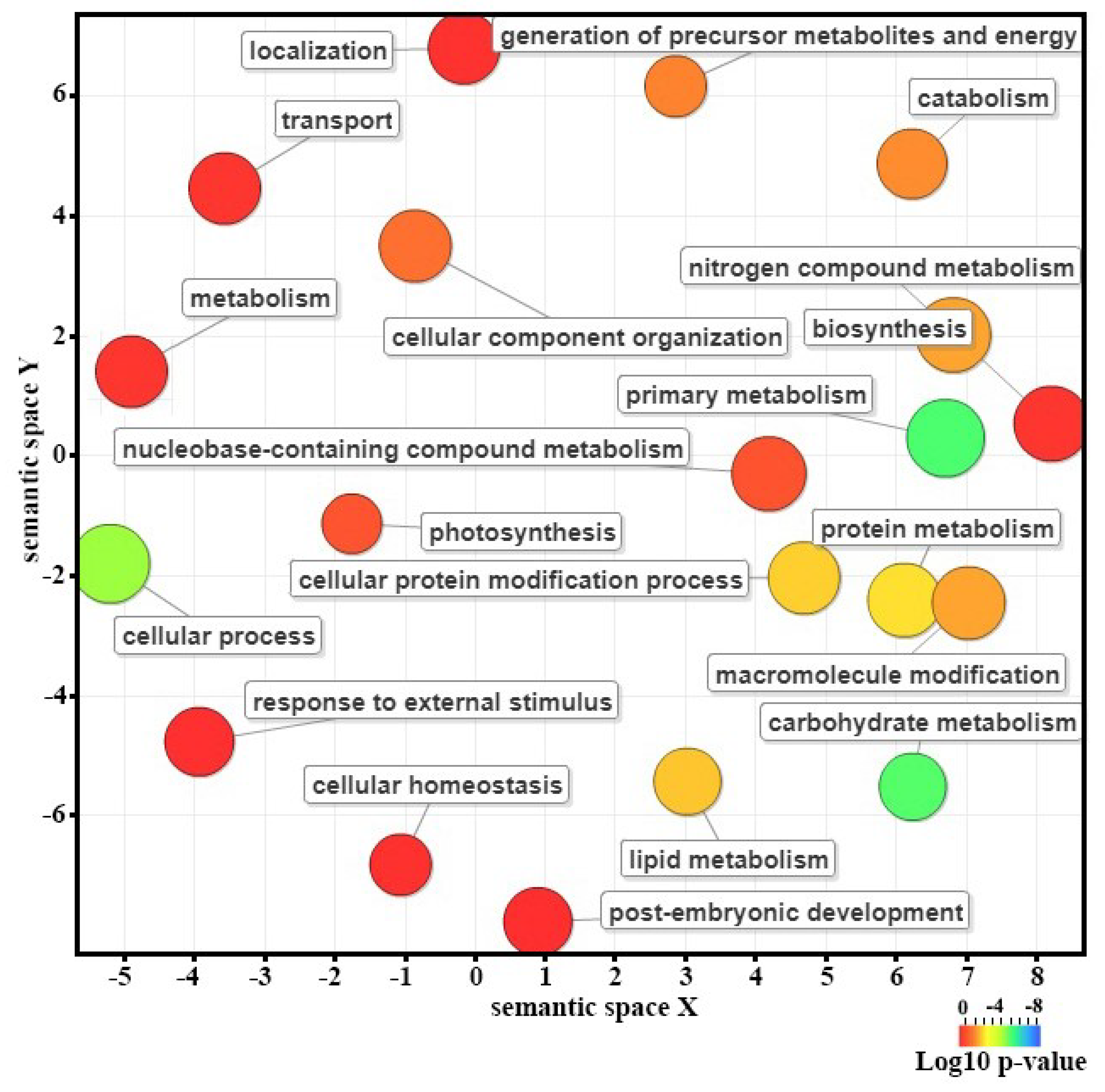
2.4. Functional Annotation and Gene Enrichment Analysis
2.5. Validation of Differentially Expressed Genes by qRT-PCR
3. Results
3.1. Biolistic Inoculation of Hop with cDNA of CBCVd and Infectivity Confirmation
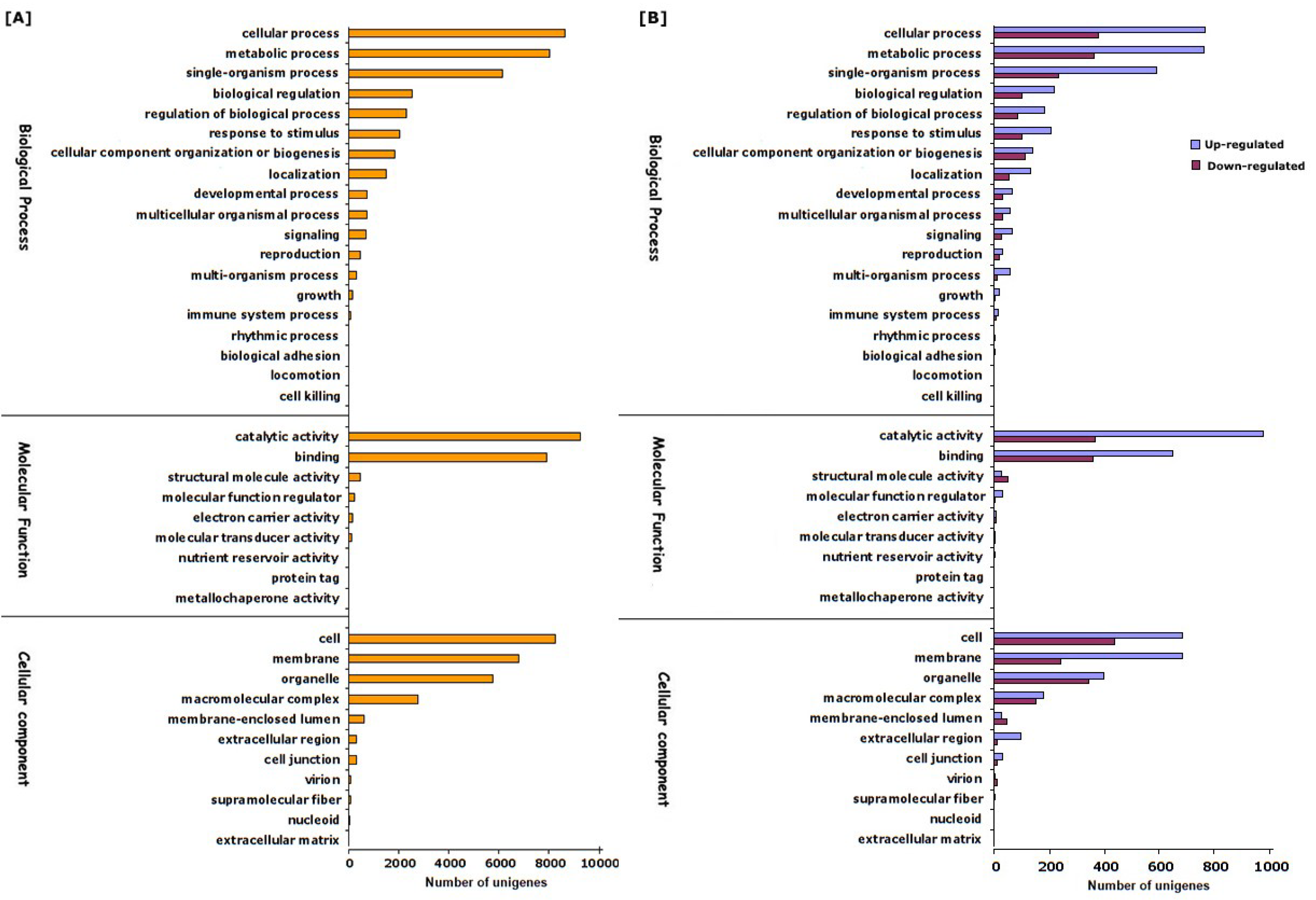
3.2. Illumina Sequencing, De Novo Assembly and Functional Annotation of Unigenes
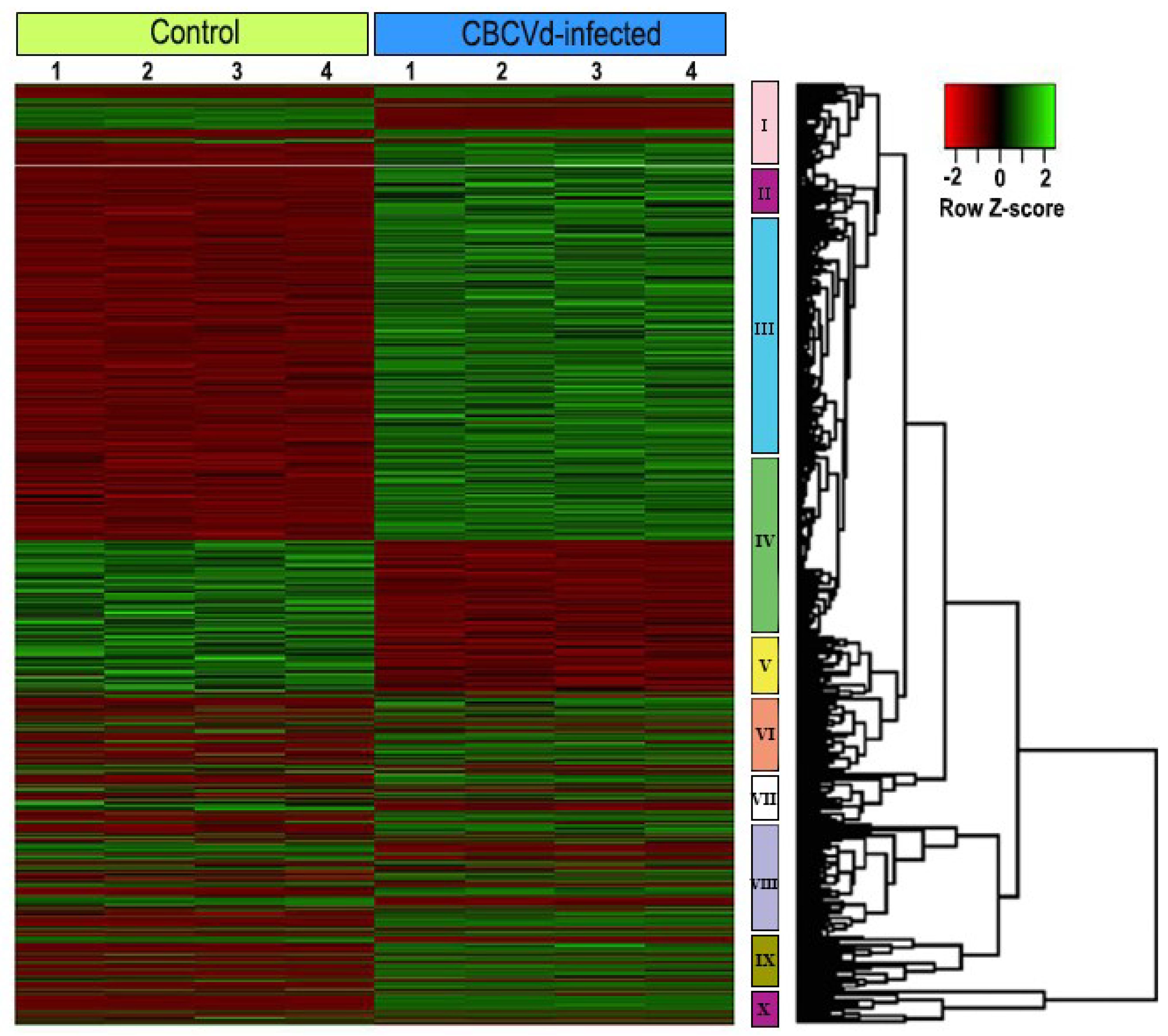
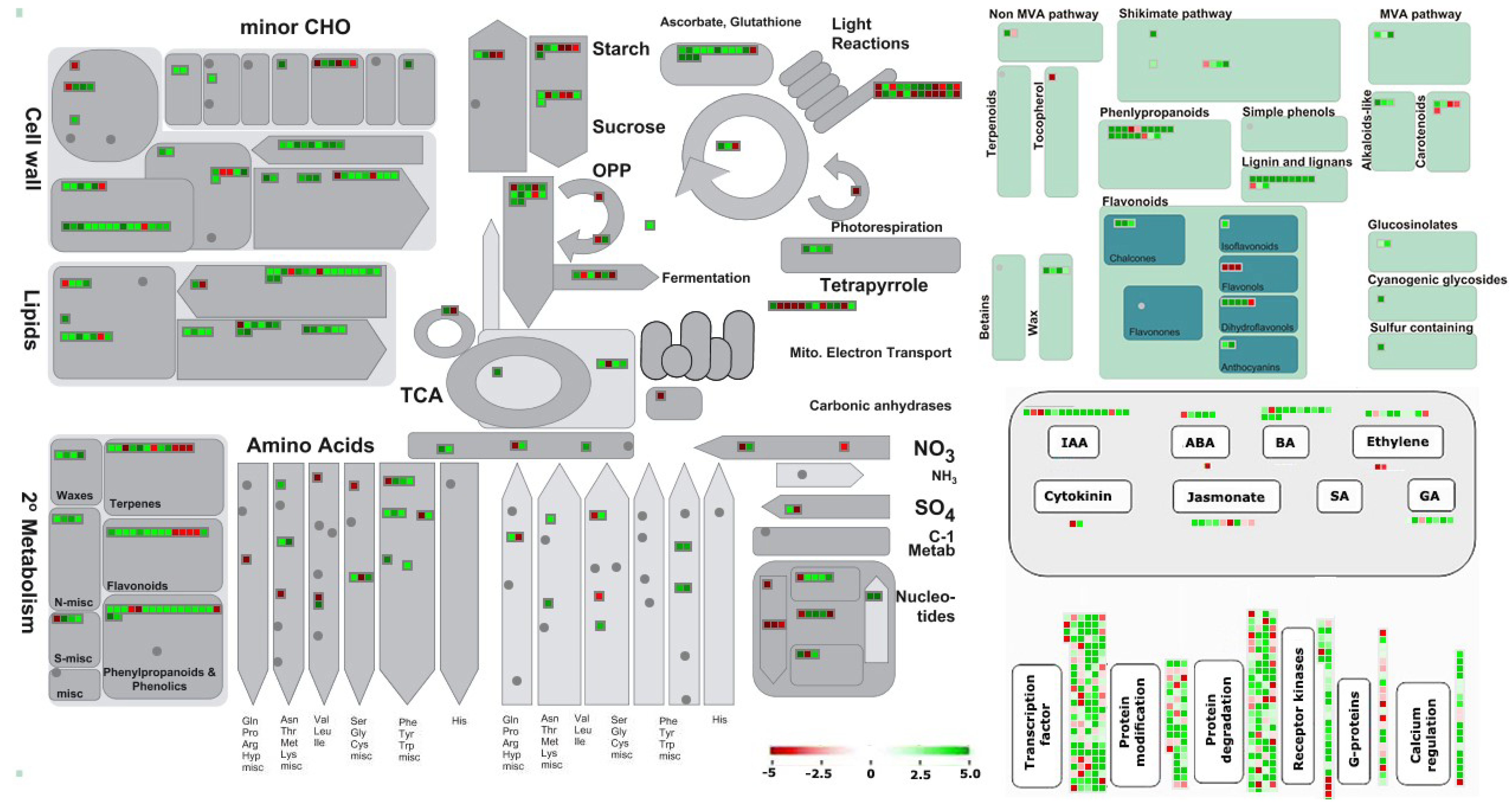
3.3. Identification and Functional Classification of Differentially Expressed Genes
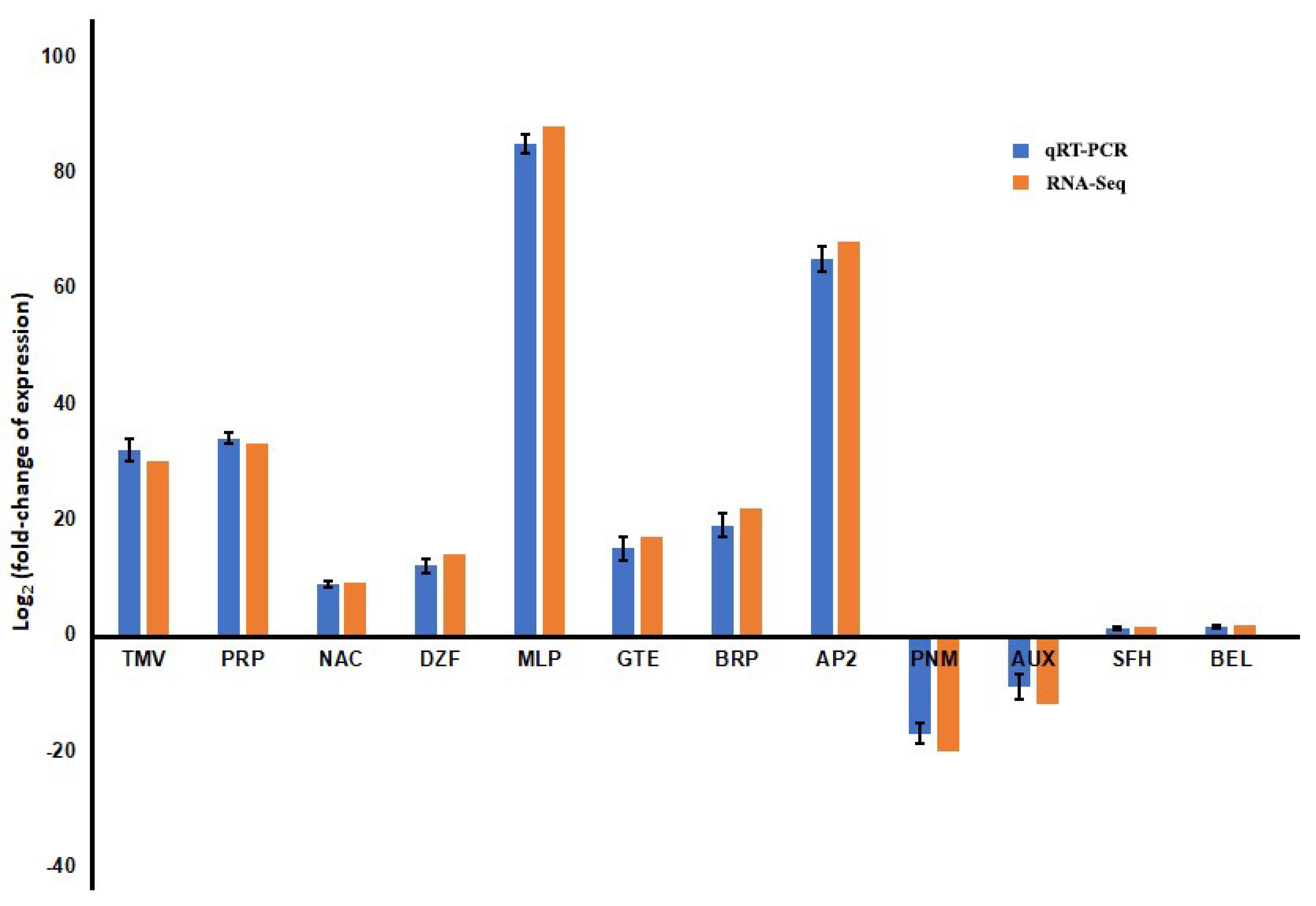
3.4. Validation of Expression Pattern of Candidate DEGs by qRT-PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diener, T.O. Discovering viroids—A personal perspective. Nat. Rev. Microbiol. 2003, 1, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Delgado, S.; Gas, M.E.; Carbonell, A.; Molina, D.; Gago, S.; de la Peña, M. Viroids: The minimal non-coding RNAs with autonomous replication. FEBS Lett. 2004, 567, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Gómez, G.; Pallás, V. A long-distance translocatable phloem protein from cucumber forms a ribonucleoprotein complex in vivo with hop stunt viroid RNA. J. Virol. 2004, 78, 10104–10110. [Google Scholar] [CrossRef] [PubMed]
- Ding, B. Viroids: Self-replicating, mobile, and fast-evolving noncoding regulatory RNAs. Wiley Interdiscip. Rev. RNA 2010, 1, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Diener, T.O. Biological properties. In The Viroids; Plenum: New York, NY, USA, 1987; Chapter 1; pp. 9–35. [Google Scholar]
- Pokorn, T.; Radišek, S.; Javornik, B.; Štajner, N.; Jakše, J. Development of hop transcriptome to support research into host-viroid interactions. PLoS ONE 2017, 12, e0184528. [Google Scholar] [CrossRef] [PubMed]
- Di Serio, F.; Flores, R.; Verhoeven, J.T.J.; Li, S.-F.; Pallás, V.; Randles, W.J.; Sano, T.; Vidalakis, G.; Owens, R.A. Current status of viroid taxonomy. Arch. Virol. 2014, 159, 3467–3478. [Google Scholar] [CrossRef] [PubMed]
- Tsagris, E.M.; de Alba Martínez, A.E.; Gozmanova, M.; Kalantidis, K. Viroids. Cell. Microbiol. 2008, 10, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Gas, M.E.; Hernández, C.; Flores, R.; Daròs, J.A. Processing of nuclear viroids in vivo: An interplay between RNA conformations. PLoS Pathog. 2007, 3, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ding, B. Inhibition of cell growth and shoot development by a specific nucleotide sequence in a noncoding viroid RNA. Plant Cell 2003, 15, 1360–1374. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Use, tolerance and avoidance of amplified RNA silencing by plants. Trends Plant Sci. 2008, 13, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Nagel, J.; Culley, L.K.; Lu, Y.; Liu, E.; Matthews, P.D.; Stevens, J.F.; Page, J.E. EST analysis of hop glandular trichomes identifies an O-methyltransferase that catalyzes the biosynthesis of xanthohumol. Plant Cell 2008, 20, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, P.; Zavatti, M. Pharmacognostic and pharmacological profile of Humulus lupulus L. J. Ethnopharmacol. 2008, 116, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Duraisamy, G.S.; Khare, M.; Kocábek, T.; Jakse, J.; Bříza, J.; Patzak, J.; Sano, T.; Matoušek, J. Genome-wide transcriptome profiling of transgenic hop (Humulus lupulus) constitutively overexpressing HlWRKY1 and HlWDR1 transcription factors. BMC Genom. 2018, 19, 739. [Google Scholar] [CrossRef] [PubMed]
- Van Cleemput, M.; Cattoor, K.; de Bosscher, K.; Haegeman, G.; de Keu-keleire, D.; Heyerick, A. Hop (Humulus lupulus)-derived bitter acids as multipotent bioactive compounds. J. Nat. Prod. 2009, 72, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.; Donaire, L.; Llave, C.; Pallas, V.; Gomez, G. High-throughput sequencing of Hop stunt viroid-derived small RNAs from cucumber leaves and phloem. Mol. Plant Pathol. 2010, 11, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Jakse, J.; Radisek, S.; Pokorn, T.; Matousek, J.; Javornik, B. Deep-sequencing revealed Citrus bark cracking viroid (CBCVd) as a highly aggressive pathogen on hop. Plant Pathol. 2015, 64, 831–842. [Google Scholar] [CrossRef]
- Mishra, A.K.; Duraisamy, G.S.; Matoušekm, J.; Radisek, S.; Javornik, B.; Jakse, J. Identification and characterization of microRNAs in Humulus lupulus using high-throughput sequencing and their response to Citrus bark cracking viroid (CBCVd) infection. BMC Genom. 2016, 17, 919. [Google Scholar] [CrossRef] [PubMed]
- Tessitori, M.; Maria, G.; Capasso, C.; Catara, G.; Rizza, S.; de Luca, V.; Catara, A.; Capasso, A.; Carginale, V. Differential display analysis of gene expression in Etrog citron leaves infected by citrus viroid III. Biochim. Biophys. Acta 2007, 1769, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Conesa, A.; Juarez, J.; Catara, A.; Navarro, L.; Duran-Vila, N.; Ancillo, G. Microarray analysis of etrog citron (Citrus medica L.) reveals changes in chloroplast, cell wall, peroxidase and symporter activities in response to viroid infection. Mol. Plant Pathol. 2012, 13, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Więsyk, A.; Iwanicka-Nowicka, R.; Fogtman, A.; Zagórski-Ostoja, W.; Góra-Sochacka, A. Time-Course Microarray Analysis Reveals Differences between Transcriptional Changes in Tomato Leaves Triggered by Mild and Severe Variants of Potato Spindle Tuber Viroid. Viruses 2018, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Marguerat, S.; Bahler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.A.; Tech, K.B.; Shao, J.Y.; Sano, T.; Baker, C.J. Global analysis of tomato gene expression during potato spindle tuber viroid infection reveals a complex array of changes affecting hormone signaling. Mol. Plant Microbe Interact. 2012, 25, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, K.; Wu, Y.; Zhang, R.; Bonar, N.; Morris, J.; Hedley, P.E.; Bryan, G.J.; Kalantidis, K.; Hornyik, C. Insight on genes affecting tuber development in potato upon potato spindle tuber viroid (PSTVd) infection. PLoS ONE 2016, 11, e0150711. [Google Scholar] [CrossRef] [PubMed]
- Herranz, M.C.; Niehl, A.; Rosales, M.; Fiore, N.; Zamorano, A.; Granell, A.; Pallas, V. A remarkable synergistic effect at the transcriptomic level in peach fruits doubly infected by prunus necrotic ringspot virus and peach latent mosaic viroid. Virol. J. 2013, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Kappagantu, M.; Bullock, J.M.; Nelson, M.E.; Eastwell, K.C. Hop stunt viroid: Effect on host (Humulus lupulus) transcriptome and its interactions with hop powdery mildew (Podospheara macularis). Mol. Plant Microbe Interact. 2017, 30, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Li, S.; Hou, W.; Fan, Z.; Xiao, H.; Lu, M.; Sano, T.; Zhang, Z. Global Transcriptomic Changes Induced by Infection of Cucumber (Cucumis sativus L.) with Mild and Severe Variants of Hop Stunt Viroid. Front Microbiol. 2017, 12, 2427. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Stehlik, J.; Procházková, J.; Orctová, L.; Wullenweber, J.; Füssy, Z.; Kováčik, J.; Duraisamy, G.; Ziegler, A.; Schubert, J.; et al. Biological and molecular analysis of the pathogenic variant C3 of potato spindle tuber viroid (PSTVd) evolved during adaptation tochamomilla (Matricaria chamomilla). Biol. Chem. 2012, 393, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Orctová, L.; Steger, G.; Riesner, D. Biolistic inoculation of plants with viroid nucleic acids. J. Virol. Methods 2004, 122, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Siglová, K.; Jakše, J.; Radišek, S.; Brass, J.R.J.; Tsushima, T.; Guček, T.; Duraisamy, G.S.; Sano, T.; Steger, G. Propagation and some physiological effects of Citrus bark cracking viroid and Apple fruit crinkle viroid in multiple infected hop (Humulus lupulus L.). J. Plant Physiol. 2017, 213, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Junker, V.; Vrba, L.; Schubert, J.; Patzak, J.; Steger, G. Molecular characterization and genome organization of 7SL RNA genes from hop (Humulus lupulus L.). Gene 1999, 239, 173–183. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Advanced heat map and clustering analysis using heatmap3. Biomed. Res. Int. 2014, 2014, 986048. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.H.; Su, Z. AgriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. “REVIGO summarizes and visualizes long lists of Gene Ontology terms”. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Deng, X.; Joshi, T.; Xu, D.; Stacey, G.; Cheng, J. Reconstructing differentially co-expressed gene modules and regulatory networks of soybean cells. BMC Genom. 2012, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Maloukh, L.; Matousek, J.; van-Bockstaele, E.; Roldán-Ruiz, I. Housekeeping gene selection for real time-PCR normalization in female hop (Humulus lupulus L.) tissues. J. Plant Biochem. Biotechnol. 2009, 18, 53–58. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, J.; Zhao, S.; Hou, Y.; Li, F.; Tai, Y.; Wan, X.; Wei, C. Transcriptome profiling using single-molecule direct RNA sequencing approach for in-depth understanding of genes in secondary metabolism pathways of Camellia sinensis. Front. Plant Sci. 2017, 8, 1205. [Google Scholar] [CrossRef] [PubMed]
- Bedre, R.; Mangu, V.R.; Srivastava, S.; Sanchez, L.E.; Baisakh, N. Transcriptome analysis of smooth cordgrass (Spartina alterniflora Loisel), a monocot halophyte, reveals candidate genes involved in its adaptation to salinity. BMC Genom. 2016, 17, 657. [Google Scholar] [CrossRef] [PubMed]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 30, 312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, H.; Chen, M.; Xiong, J.; Cai, H.; Liu, Y. Transcriptome analysis reveals potential genes involved in flower pigmentation in a red-flowered mutant of white clover (Trifolium repens L.). Genomics 2018, 110, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jiao, C.; Zheng, Y.; Sun, H.; Liu, W.; Cai, X.; Wang, X.; Liu, S.; Xu, Y.; Mou, B.; et al. De novo and comparative transcriptome analysis of cultivated and wild spinach. Sci. Rep. 2015, 5, 17706. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Takahashi, H.; Nakamura, M.; Bunsupa, S.; Yoshimoto, N.; Yamamoto, H.; Suzuki, H.; Shibata, D.; Yamazaki, M.; Saito, K. Transcriptome analysis of nine tissues to discover genes involved in the biosynthesis of active ingredients in Sophora flavescens. Biol. Pharm. Bull. 2015, 38, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.D.; Ramos-gonzález, P.L.; Nunes, M.A. Citrus leprosis virus C infection results in hypersensitive-like response, suppression of the JA/ET plant defense pathway and promotion of the colonization of its mite vector. Front. Plant Sci. 2016, 7, 1757. [Google Scholar] [CrossRef] [PubMed]
- Camejo, D.; Guzmán-Cedeño, Á.; Moreno, A. Reactive Oxygen Species, essential molecules, during plant-pathogen interactions. Plant Physiol. Biochem. 2016, 103, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Aliferis, K.A.; Faubert, D.; Jabaji, S. A metabolic profiling strategy for the dissection of plant defense against fungal pathogens. PLoS ONE 2014, 9, e111930. [Google Scholar] [CrossRef] [PubMed]
- Semancik, J.S.; Vidalakis, G. The question of Citrus viroid IV as a Cocadviroid. Arch. Virol. 2005, 150, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Spoel, S.H.; Dong, X. How do plants achieve immunity? Defence without specialized immune cells. Nat. Rev. Immunol. 2012, 12, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Gao, X.; Feng, B.; Sheen, J.; Shan, L.; He, P. Plant immune response to pathogens differs with changing temperatures. Nat. Commun. 2013, 4, 2530. [Google Scholar] [CrossRef] [PubMed]
- Ucci, J.W.; Kobayashi, Y.; Choi, G.; Alexandrescu, A.T.; Cole, J.L. Mechanism of interaction of the double-stranded RNA (dsRNA) binding domain of protein kinase R with short dsRNA sequences. Biochemistry 2007, 46, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Bilgin, D.D.; Liu, Y.; Schiff, M.; Dinesh-Kumar, S.P. P58IPK, a plant ortholog of double-stranded RNA-dependent protein kinase PKR inhibitor, functions in viral pathogenesis. Dev. Cell 2003, 4, 651–661. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Chakraborty, S. Chloroplast: The Trojan horse in plant-virus interaction. Mol. Plant Pathol. 2018, 19, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Nohales, M.; Molina-Serrano, D.; Flores, R.; Daròs, J.A. Involvement of the chloroplastic isoform of tRNA ligase in the replication of viroids belonging to the family Avsunviroidae. J. Virol. 2012, 86, 8269–8276. [Google Scholar] [CrossRef] [PubMed]
- Shu, K.; Liu, X.D.; Xie, Q.; He, Z.H. Two faces of one seed: Hormonal regulation of dormancy and germination. Mol. Plant 2016, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Novak, O.; Napier, R.; Ljung, K. Zooming in on plant hormone analysis: Tissue- and cell-specific approaches. Annu. Rev. Plant Biol. 2017, 68, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Collum, T.D.; Culver, J.N. The impact of phytohormones on virus infection and disease. Curr. Opin. Virol. 2016, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, Y.; Ding, B.; Fei, Z.J. Comprehensive transcriptome analyses reveal that potato spindle tuber viroid triggers genome-wide changes in alternative splicing, inducible trans-acting activity of phasiRNAs and immune responses. J. Virol. 2017, 91, e00247-17. [Google Scholar] [CrossRef] [PubMed]
- López-Gresa, M.P.; Lisón, P.; Yenush, L.; Conejero, V.; Rodrigo, I.; Bellés, J.M. Salicylic acid is involved in the basal resistance of tomato plants to citrus exocortis viroid and tomato spotted wilt virus. PLoS ONE 2016, 11, e0166938. [Google Scholar] [CrossRef] [PubMed]
- Verchot, J. The ER quality control and ER associated degradation machineries are vital for viral pathogenesis. Front. Plant Sci. 2014, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Verchot, J. Plant Virus Infection and the Ubiquitin Proteasome Machinery: Arms Race along the Endoplasmic Reticulum. Viruses 2016, 8, e314. [Google Scholar] [CrossRef] [PubMed]
- Sasvari, Z.; Alatriste Gonzalez, P.; Nagy, P.D. Tombusvirus-yeast interactions identify conserved cell-intrinsic viral restriction factors. Front. Plant Sci. 2014, 5, 383. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Gisel, A.; Rodio, M.E.; Degado, S.; Flores, R.; di Serio, F. Small RNAs containing the pathogenic determinant of a chloroplast-replicating viroid guide the degradation of a host mRNA as predicted by RNA silencing. Plant J. 2012, 70, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.B.; Rochon, D. Cucumber Necrosis Virus Recruits Cellular Heat Shock Protein 70 Homologs at Several Stages of Infection. J. Virol. 2015, 90, 3302–3317. [Google Scholar] [CrossRef] [PubMed]
- Hafren, A.; Hofius, D.; Ronnholm, G.; Sonnewald, U.; Makinen, K. HSP70 and its cochaperone CPIP promote potyvirus infection in Nicotiana benthamiana by regulating viral coat protein functions. Plant Cell 2010, 22, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.S.; Dadalto, S.P.; Gonçalves, A.B.; de Souza, G.B.; Barros, V.A.; Fietto, L.G. Transcription factor functional protein-protein interactions in plant defense responses. Proteomes 2014, 2, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Kocábek, T.; Patzak, J.; Bříza, J.; Siglová, K.; Mishra, A.K.; Duraisamy, G.S.; Týcová, A.; Ono, E.; Krofta, K. The “putative” role of transcription factors from HlWRKY family in the regulation of the final steps of prenylflavonid and bitter acids biosynthesis in hop (Humulus lupulus L.). Plant Mol. Biol. 2016, 92, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front Sci. 2012, 3, 222. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Library | Number | Total Bases (GB) |
|---|---|---|---|
| Raw read | MI | 33,752,449 | 3.21 |
| CI | 40,023,124 | 3.60 | |
| Clean read | MI | 24,399,800 | 1.79 |
| CI | 36,686,240 | 2.72 | |
| Average Length (bp) | MI | 421 | |
| CI | 418 | ||
| Unigenes | |||
| No. of Unigenes (n) | 27,904 | ||
| Average Length (bp) | 451 | ||
| Maximum Length (bp) | 2590 | ||
| Minimum Length (bp) | 90 |
| KEGG Categories | Number of | KEGG Categories | Number of | ||||
|---|---|---|---|---|---|---|---|
| UG | UR | DR | UG | UR | DR | ||
| Metabolism | Organismal System | ||||||
| Carbohydrate Metabolism | 1457 | 168 | 54 | Immune system | 650 | 24 | 23 |
| Energy metabolism | 608 | 58 | 47 | Endocrine system | 692 | 66 | 29 |
| Lipid metabolism | 768 | 149 | 41 | Circulatory system | 93 | 8 | 3 |
| Nucleotide metabolism | 320 | 19 | 29 | Digestive system | 167 | 18 | 6 |
| Amino acid metabolism | 841 | 126 | 50 | Excretory system | 111 | 12 | 2 |
| Metabolism of other amino acids | 307 | 44 | 24 | Nervous system | 495 | 44 | 13 |
| Glycan biosynthesis and metabolism | 299 | 43 | 10 | Sensory system | 49 | 11 | 3 |
| Metabolism of cofactors and vitamins | 412 | 41 | 13 | Development | 74 | 5 | 6 |
| Metabolism of terpenoids and polyketides | 222 | 31 | 26 | Aging | 219 | 10 | 17 |
| Biosynthesis of other secondary metabolites | 453 | 110 | 10 | Environmental adaptation | 506 | 55 | 26 |
| Xenobiotics biodegradation and metabolism | 239 | 61 | 34 | ||||
| Enzyme families | 921 | 106 | 41 | ||||
| Genetic information processing | |||||||
| Transcription | 316 | 71 | 60 | ||||
| Translation | 846 | 111 | 235 | ||||
| Folding, sorting and degradation | 730 | 201 | 65 | ||||
| Replication and repair | 271 | 63 | 60 | ||||
| RNA family | 0 | 0 | 0 | ||||
| Cellular Process | |||||||
| Transport and catabolism | 916 | 174 | 52 | Unclassified | |||
| Cell growth and death | 684 | 49 | 22 | Metabolism | 2007 | 44 | 11 |
| Cellular community—eukaryotes | 178 | 7 | 3 | Genetic information processing | 5896 | 6 | 5 |
| Cellular community—prokaryotes | 113 | 20 | 3 | Cellular processes and signaling | 1833 | 8 | 3 |
| Cell motility | 55 | 14 | 14 | Viral protein family | 0 | 0 | 0 |
| Environmental information processing | Poorly characterized | 585 | 8 | 6 | |||
| Membrane transport | 64 | 96 | 33 | ||||
| Signal transduction | 2134 | 165 | 63 | ||||
| Signaling molecules and interaction | 187 | 26 | 7 | ||||
| Total | 26,718 | 2272 | 1149 | ||||
| GO ID | Ontology | Category | Number of DEGs | Number of Unigenes in Subgroup | FDR | p-value |
|---|---|---|---|---|---|---|
| GO:0008152 | metabolic process | P | 1008 | 10,614 | 7.10 × 10−92 | 2.20 × 10−94 |
| GO:0044238 | primary metabolic process | P | 816 | 8995 | 9.20 × 10−59 | 5.60 × 10−61 |
| GO:0005975 | carbohydrate metabolic process | P | 199 | 866 | 5.00 × 10−58 | 4.60 × 10−60 |
| GO:0009987 | cellular process | P | 949 | 11,684 | 2.70 × 10−50 | 3.30 × 10−52 |
| GO:0019538 | protein metabolic process | P | 388 | 4009 | 3.80 × 10−28 | 5.80 × 10−30 |
| GO:0006464 | protein modification process | P | 193 | 1474 | 3.70 × 10−26 | 6.70 × 10−28 |
| GO:0006629 | lipid metabolic process | P | 135 | 841 | 3.40 × 10−25 | 7.30 × 10−27 |
| GO:0009058 | biosynthetic process | P | 435 | 5118 | 2.50 × 10−21 | 6.80 × 10−23 |
| GO:0009056 | catabolic process | P | 159 | 1307 | 1.00 × 10−18 | 3.10 × 10−20 |
| GO:0015979 | photosynthesis | P | 34 | 162 | 5.80 × 10−9 | 2.30 × 10−10 |
| GO:0006810 | transport | P | 136 | 1846 | 0.0025 | 0.00012 |
| GO:0051179 | localization | P | 136 | 1922 | 9.90 × 10−3 | 0.00058 |
| GO:0019725 | cellular homeostasis | P | 21 | 174 | 1.30 × 10−2 | 0.00082 |
| GO:0009605 | response to external stimulus | P | 39 | 429 | 2.20 × 10−2 | 0.0014 |
| GO:0003824 | catalytic activity | F | 1145 | 9638 | 3.60 × 10−18 | 4.10 × 10−189 |
| GO:0016740 | transferase activity | F | 360 | 3321 | 6.90 × 10−35 | 1.60 × 10−36 |
| GO:0016787 | hydrolase activity | F | 370 | 3468 | 8.10 × 10−35 | 2.80 × 10−36 |
| GO:0005488 | binding | F | 859 | 11,258 | 1.50 × 10−33 | 8.60 × 10−35 |
| GO:0016301 | kinase activity | F | 140 | 1641 | 1.20 × 10−06 | 8.50 × 10−8 |
| GO:0016772 | transferase activity, transferring phosphorus-containing groups | F | 140 | 1887 | 7.70 × 10−4 | 7.10 × 10−5 |
| GO:0005215 | transporter activity | F | 108 | 1477 | 7.10 × 10−3 | 0.00075 |
| GO:0008135 | translation factor activity, nucleic acid binding | F | 21 | 181 | 1.10 × 10−2 | 0.0013 |
| GO:0005198 | structural molecule activity | F | 54 | 659 | 1.40 × 10−2 | 0.0018 |
| GO:0016020 | membrane | C | 426 | 4068 | 2.80 × 10−38 | 1.60 × 10−40 |
| GO:0005737 | cytoplasm | C | 582 | 6822 | 1.30 × 10−30 | 2.10 × 10−32 |
| GO:0005623 | cell | C | 1015 | 15,217 | 7.40 × 10−20 | 2.90 × 10−21 |
| GO:0043226 | organelle | C | 594 | 8155 | 5.60 × 10−16 | 3.20 × 10−17 |
| GO:0005840 | ribosome | C | 79 | 524 | 1.10 × 10−13 | 6.80 × 10−15 |
| GO:0009536 | plastid | C | 227 | 2965 | 3.20 × 10−7 | 3.80 × 10−8 |
| GO:0005618 | cell wall | C | 49 | 403 | 3.00 × 10−6 | 3.80 × 10−7 |
| GO:0005829 | cytosol | C | 84 | 912 | 1.80 × 10−5 | 2.50 × 10−6 |
| GO:0005886 | plasma membrane | C | 100 | 1456 | 0.041 | 0.0064 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Kumar, A.; Mishra, D.; Nath, V.S.; Jakše, J.; Kocábek, T.; Killi, U.K.; Morina, F.; Matoušek, J. Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.). Viruses 2018, 10, 570. https://doi.org/10.3390/v10100570
Mishra AK, Kumar A, Mishra D, Nath VS, Jakše J, Kocábek T, Killi UK, Morina F, Matoušek J. Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.). Viruses. 2018; 10(10):570. https://doi.org/10.3390/v10100570
Chicago/Turabian StyleMishra, Ajay Kumar, Atul Kumar, Deepti Mishra, Vishnu Sukumari Nath, Jernej Jakše, Tomáš Kocábek, Uday Kumar Killi, Filis Morina, and Jaroslav Matoušek. 2018. "Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.)" Viruses 10, no. 10: 570. https://doi.org/10.3390/v10100570
APA StyleMishra, A. K., Kumar, A., Mishra, D., Nath, V. S., Jakše, J., Kocábek, T., Killi, U. K., Morina, F., & Matoušek, J. (2018). Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.). Viruses, 10(10), 570. https://doi.org/10.3390/v10100570