Spontaneous and Engineered Compensatory HSV Mutants that Counteract the Host Antiviral PKR Response

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PKR Innate Antiviral Response
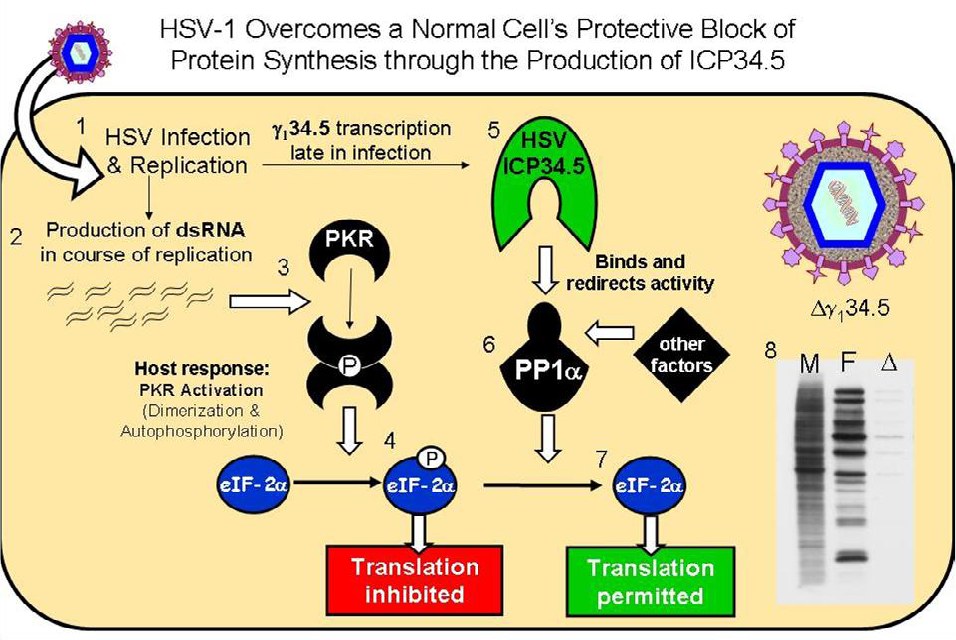
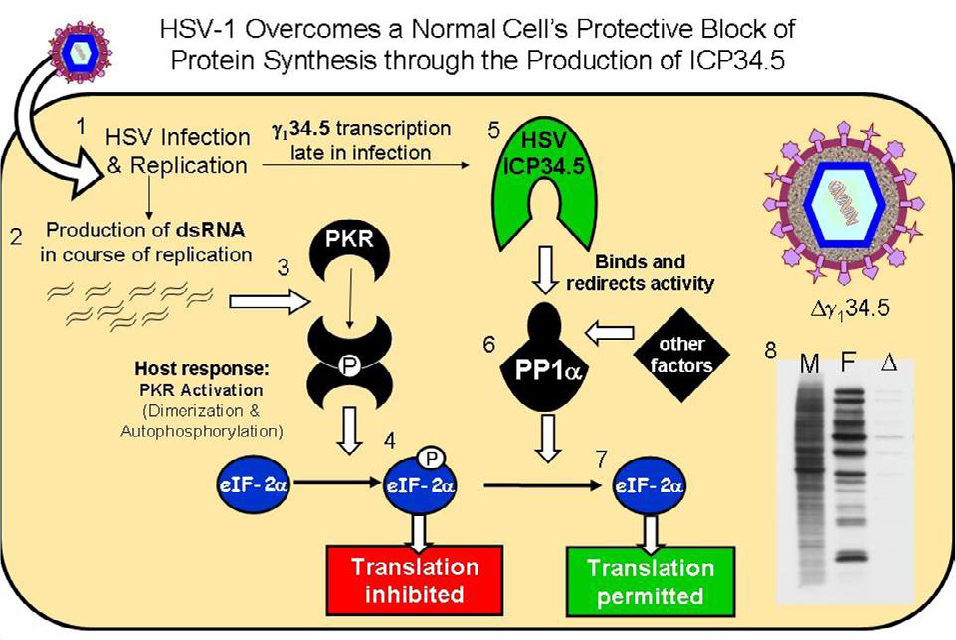
3. The HSV-1 γ134.5 Gene
4.γ134.5 HSV as Oncolytic Vectors

5. Serial Passage and Escape Mutations
5.1. In vitro passage
5.2. In vivo passage
6. Engineered Mutations
6.1. R8309
6.2. GΔ47 – engineering the ΔUS12 αUS11 mutation
6.3. Controlled expression of the γ134.5 gene or tumor targeting of γ134.5 containing HSV
6.4. Chimeric HSV
7. Conclusions
Acknowledgments
References and Notes
- Samuel, C.E. Antiviral actions of interferons . Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Katze, M. G. Regulation of the interferon-induced PKR: can viruses cope? Trends Microbiol. 1995, 3, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Thomis, D.C.; Samuel, C.E. Mechanism of interferon action: evidence for intermolecular autophosphorylation and autoactivation of the interferon-induced, RNA-dependent protein kinase PKR. J. Virol. 1993, 67, 7695–7700. [Google Scholar] [PubMed]
- Ortega, L.G.; McCotter, M.D.; Henry, G.L.; McCormack, S.J.; Thomis, D.C.; Samuel, C.E. Mechanism of interferon action. Biochemical and genetic evidence for the intermolecular association of the RNA-dependent protein kinase PKR from human cells. Virology 1996, 215, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Signal integration via PKR . Sci. STKE 2001, 89, RE2. [Google Scholar]
- Smith, K.D.; Mezhir, J.J.; Bickenbach, K.; Veerapong, J.; Charron, J.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of herpes simplex virus 1 . J. Virol. 2006, 80, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Whitmore, M.; Xu, Z.; Jiang, Z.; Li, X.; Williams, B.R. Protein kinase R (PKR) interacts with and activates mitogen-activated protein kinase kinase 6 (MKK6) in response to double-stranded RNA stimulation. J. Biol. Chem. 2004, 279, 37670–37676. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway . Proc. Natl. Acad. Sci. U S A 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.L.; Johnson, E.M. Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J. Cell Biol. 1998, 142, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. PKR: a sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Chen, J.J.; Gross, M.; Roizman, B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1 . Proc. Natl. Acad. Sci. U S A 1995, 92, 10516–10520. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Kern. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [PubMed]
- He, B.; Chou, J.; Liebermann, D.A.; Hoffman, B.; Roizman, B. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J. Virol. 1996, 70, 84–90. [Google Scholar] [PubMed]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U S A 1997, 94, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Lord, K.A.; Alamo, I.; Hollander, M.C.; Carrier, F.; Ron, D.; Kohn, K.W.; Hoffman, B.; Liebermann, D.A.; Fornace, A.J. CarrierF.RonD.KohnK.W.HoffmanB.LiebermannD.A.FornaceA.J.The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that synergistically suppress cell growth . Mol. Cell Biol. 1994, 14, 2361–2371. [Google Scholar] [PubMed]
- Chou, J.; Roizman, B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells . Proc. Natl. Acad. Sci. U S A 1992, 89, 3226–3270. [Google Scholar]
- Cheng, G.; Yang, K.; He, B. Dephosphorylation of eIF-2alpha mediated by the gamma(1)34.5 protein of herpes simplex virus type 1 is required for viral response to interferon but is not sufficient for efficient viral replication . J. Virol. 2003, 77, 10154–10161. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A.; Gross, M.; Gillespie, G.Y.; Roizman, B. Second-site mutation outside of the U(S)10-12 domain of Deltagamma(1)34.5 herpes simplex virus 1 recombinant blocks the shutoff of protein synthesis induced by activated protein kinase R and partially restores neurovirulence . J. Virol. 2002, 76, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Harle, P.; Cull, V.; Agbaga, M.P.; Silverman, R.; Williams, B.R.; James, C.; Carr, D.J. Differential effect of murine alpha/beta interferon transgenes on antagonization of herpes simplex virus type 1 replication. J. Virol. 2002, 76, 6558–6567. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yokosawa, N.; Okabayashi, T.; Suzutani, T.; Miura, S.; Jimbow, K.; Fujii, N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 2004, 78, 6282–6286. [Google Scholar] [CrossRef] [PubMed]
- Markovitz, N.S.; Baunoch, D.; Roizman, B. The range and distribution of murine central nervous system cells infected with the gamma(1)34.5- mutant of herpes simplex virus 1 . J. Virol. 1997, 71, 5560–5569. [Google Scholar] [PubMed]
- Andreansky, S.S.; He, B.; Gillespie, G.Y.; Soroceanu, L.; Markert, J.; Chou, J.; Roizman, B.; Whitley, R.J. The application of genetically engineered herpes simplex viruses to the treatment of experimental brain tumors. Proc. Natl. Acad. Sci. U S A 1996, 93, 11313–11318. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Cobbs, C.; Markert, J. Gene Therapy of Glioma: A Review. Perspectives in Neurological Surgery 1999, in press. [Google Scholar]
- Markert, J.M.; Gillespie, G.Y.; Weichselbaum, R.R.; Roizman, B.; Whitley, R.J. Genetically engineered HSV in the treatment of glioma: a review. Rev. Med. Virol. 2000, 10, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; Mabbs, R.; Brown, M. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef]
- Bower, J.R.; Mao, H.; Durishin, C.; Rozenbom, E.; Detwiler, M.; Rempinski, D.; Karban, T.L.; Rosenthal, K.S. Intrastrain variants of herpes simplex virus type 1 isolated from a neonate with fatal disseminated infection differ in the ICP34.5 gene, glycoprotein processing, and neuroinvasiveness. J. Virol. 1999, 73, 3843–3853. [Google Scholar] [PubMed]
- Mao, H.; Rosenthal, K.S. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential . J. Virol. 2003, 77, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1 . J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Mohr, I.; Gluzman, Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. Embo J. 1996, 15, 4759–4766. [Google Scholar] [PubMed]
- Cassady, K.A.; Gross, M.; Roizman, B. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2 . J. Virol. 1998, 72, 8620–8626. [Google Scholar] [PubMed]
- Cassady, K.A.; Gross, M.; Roizman, B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J. Virol. 1998, 72, 7005–7011. [Google Scholar] [PubMed]
- Mohr, I.; Sternberg, D.; Ward, S.; Leib, D.; Mulvey, M.; Gluzman, Y. A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals . J. Virol. 2001, 75, 5189–5196. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.; MacGregor, J.; Markus, S.; Ha, S.; Mohr, I. Enhanced antitumor efficacy of a herpes simplex virus mutant isolated by genetic selection in cancer cells. Proc. Natl. Acad. Sci. U S A 2001, 98, 8804–8808. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.C.; Price, K.H.; Parker, J.N.; Samuel, S.L.; Meleth, S.; Cassady, K.A.; Gillespie, G.Y.; Whitley, R.J.; Markert, J.M. Serial passage through human glioma xenografts selects for a Deltagamma134.5 herpes simplex virus type 1 mutant that exhibits decreased neurotoxicity and prolongs survival of mice with experimental brain tumors . J. Virol. 2006, 80, 7308–7315. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. U S A 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Martuza, R.L.; Rabkin, S.D. Intracarotid delivery of oncolytic HSV vector G47Delta to metastatic breast cancer in the brain. Gene Ther. 2005, 12, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Martuza, R.L.; Rabkin, S.D.; Ito, Y.; Todo, T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin. Cancer Res. 2005, 11, 7886–7890. [Google Scholar] [CrossRef]
- Messerli, S.M.; Prabhakar, S.; Tang, Y.; Mahmood, U.; Giovannini, M.; Weissleder, R.; Bronson, R.; Martuza, R.; Rabkin, S.; Breakefield, X.O. Treatment of schwannomas with an oncolytic recombinant herpes simplex virus in murine models of neurofibromatosis type 2. Hum. Gene Ther. 2006, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.Y.; Saeki, Y.; Chiocca, E.A. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene- mediated virulence toward tumor and cycling cells . J. Virol. 1999, 73, 7556–7564. [Google Scholar] [PubMed]
- Nakamura, H.; Kasuya, H.; Mullen, J.T.; Yoon, S.S.; Pawlik, T.M.; Chandrasekhar, S.; Donahue, J.M.; Chiocca, E.A.; Chung, R.Y.; Tanabe, K.K. Regulation of herpes simplex virus gamma(1)34.5 expression and oncolysis of diffuse liver metastases by Myb34.5. J. Clin. Invest. 2002, 109, 871–882. [Google Scholar] [PubMed]
- Kanai, R.; Tomita, H.; Shinoda, A.; Takahashi, M.; Goldman, S.; Okano, H.; Kawase, T.; Yazaki, T. Enhanced therapeutic efficacy of G207 for the treatment of glioma through Musashi1 promoter retargeting of gamma34.5-mediated virulence. Gene Ther. 2006, 13, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor . Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Ye, G.J.; Debinski, W.; Roizman, B. Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc. Natl. Acad. Sci. U S A 2002, 99, 15124–15129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Roizman, B. Characterization of a recombinant herpes simplex virus 1 designed to enter cells via the IL13Ralpha2 receptor of malignant glioma cells. J. Virol. 2005, 79, 5272–5277. [Google Scholar] [CrossRef] [PubMed]
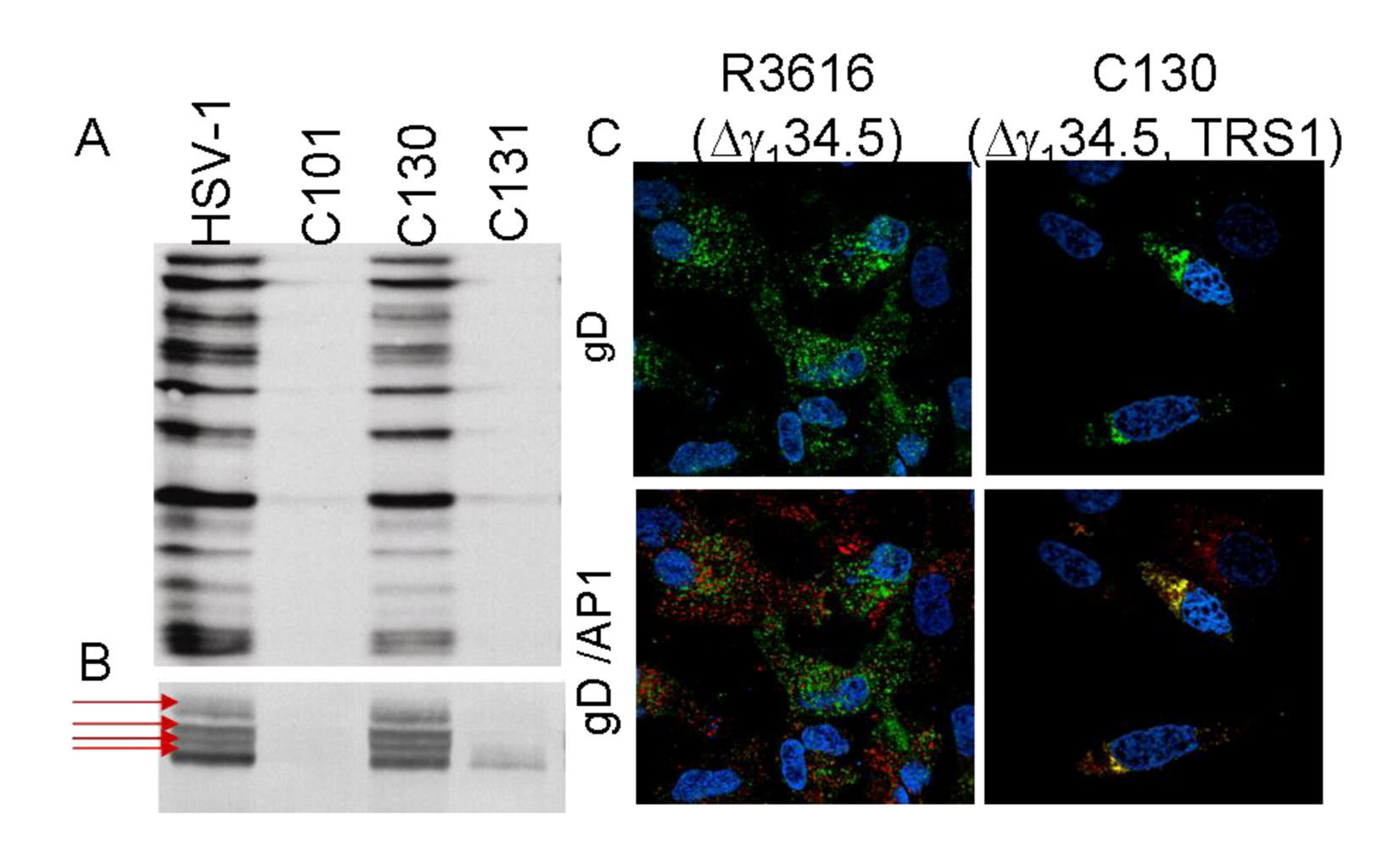
- Child, S.J.; Hakki, M.; De Niro, K.L.; Geballe, A.P. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 2004, 78, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Shah, A.C.; Parker, J.N.; Shimamura, M.; Cassady, K.A. Spontaneous and Engineered Compensatory HSV Mutants that Counteract the Host Antiviral PKR Response. Viruses 2009, 1, 510-522. https://doi.org/10.3390/v1030510
Shah AC, Parker JN, Shimamura M, Cassady KA. Spontaneous and Engineered Compensatory HSV Mutants that Counteract the Host Antiviral PKR Response. Viruses. 2009; 1(3):510-522. https://doi.org/10.3390/v1030510
Chicago/Turabian StyleShah, Amish C., Jacqueline N. Parker, Masako Shimamura, and Kevin A. Cassady. 2009. "Spontaneous and Engineered Compensatory HSV Mutants that Counteract the Host Antiviral PKR Response" Viruses 1, no. 3: 510-522. https://doi.org/10.3390/v1030510
APA StyleShah, A. C., Parker, J. N., Shimamura, M., & Cassady, K. A. (2009). Spontaneous and Engineered Compensatory HSV Mutants that Counteract the Host Antiviral PKR Response. Viruses, 1(3), 510-522. https://doi.org/10.3390/v1030510