Activation of the Antiviral Kinase PKR and Viral Countermeasures

Abstract
:1. Introduction
2. Activation of PKR by Viral Ribonucleotides
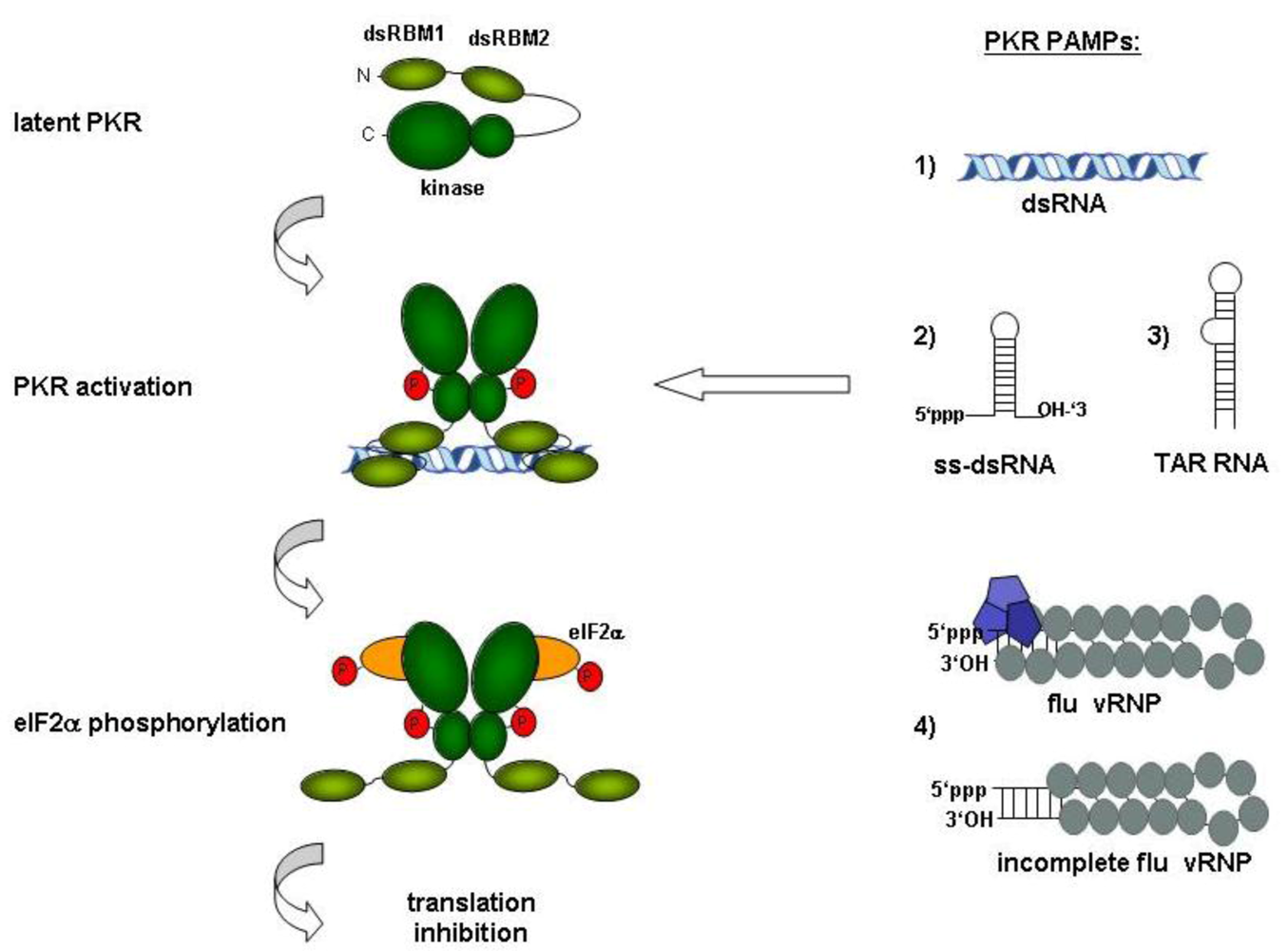
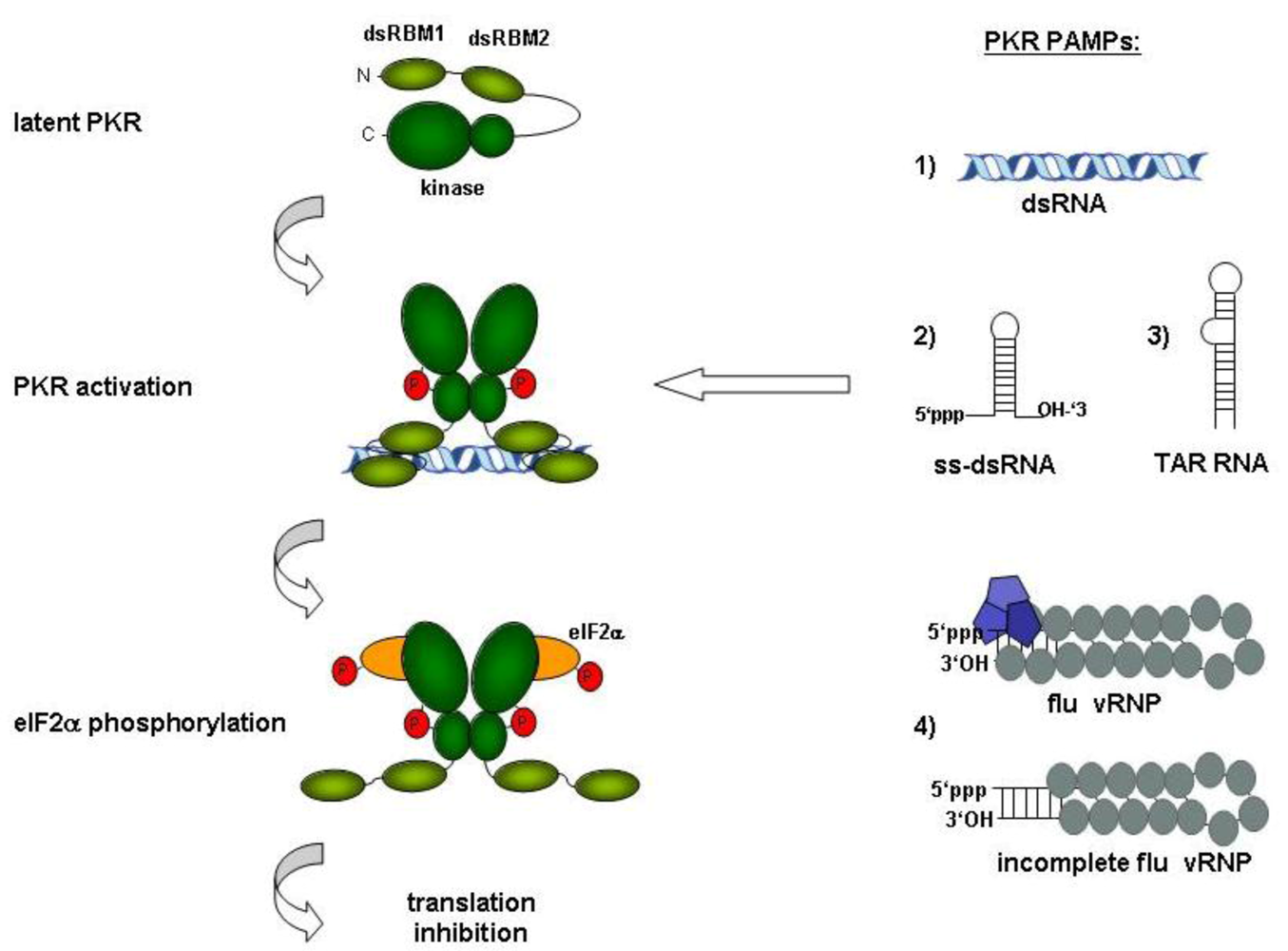
2.1. Synthetic RNAs
2.2. Viral RNAs

{kind=link}
| Virus (genome) | dsRNA detected in IFA | Origin of PKR activating RNA | Reference |
|---|---|---|---|
| VacV, AdV, HSV-1, HCMV, MCMV (DNA) | + | overlapping converging transcription | [36–41] |
| HIV-1 (RNA/DNA) | n. a. | TAR RNA/possibly as dimer | [52–54,56] |
| ReoV (dsRNA) | + | dsRNA genome | [39,42,49] |
| Rubella V., SFV, SINV, SARS CoV, EMCV, Kunjin V., PolioV, TBEV, HCV, DENV. (+ssRNA) | + | replication intermediates or base-paired secondary structure elements | [39,43–48] |
| Influenza V. (-ssRNA, segm.) | - | vRNP/complementary 3’ and 5’ termini of vRNA (flu B virus) | [28,39,55] |
| LaCrosse V. (-ssRNA, segm.) | - | panhandle structure of vRNA? | [39,76] |
| SenV (-ssRNA, non-segm.) | - | ? | [91] |
| SenV C protein mutant, NDV Ulster strain (-ssRNA, non-segm.) | + | replication intermediates | [91] |
3. Viral Countermeasures
3.1. Influenza A and B viruses
| Viral product | Virus | Mode of inhibition |
|---|---|---|
| NS1 | influenza A virus | direct interaction with PKR |
| NS | influenza B virus | dsRNA-mediated interaction with PKR |
| NSs | Rift Valley Fever virus | proteasome-mediated degradation of PKR |
| VP35 | Ebola Virus | unknown |
| pTRS1/pIRS1 | human CMV | relocalization of PKR, interaction with PKR |
| m142/m143 | murine CMV | relocalization of PKR, direct interaction with PKR |
3.2. Rift Valley fever virus
3.3. Ebola virus
3.4. Cytomegalovirus
4. Conclusions
Acknowledgments
References and Notes
- Garcia-Sastre, A.; Biron, C.A. Type 1 interferons and the virus-host relationship: A lesson in detente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Haines, G.K.; Panos, R.J.; Bak, P.M.; Brown, T.; Zielinski, M.; Leyland, J.; Radosevich, J.A. Interferon-responsive protein kinase (p68) and proliferating cell nuclear antigen are inversely distributed in head and neck squamous cell carcinoma . Tumor Biol. 1998, 19, 52–59. [Google Scholar] [CrossRef]
- Kuhen, K.L.; Samuel, C.E. Mechanism of interferon action: Functional characterization of positive and negative regulatory domains that modulate transcriptional activation of the human RNA-dependent protein kinase Pkr promoter. Virology 1999, 254, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.L. Activation of PKR: An open and shut case? Trends Biochem. Sci 2007, 32, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Galabru, J.; Hovanessian, A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem. 1987, 262, 15538–15544. [Google Scholar] [PubMed]
- Romano, P.R.; Zhang, F.; Tan, S.L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: Role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol. 1998, 18, 7304–7316. [Google Scholar] [PubMed]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Pathak, V.K.; Schindler, D.; Hershey, J.W. Generation of a mutant form of protein synthesis initiation factor eIF-2 lacking the site of phosphorylation by eIF-2 kinases. Mol. Cell. Biol. 1988, 8, 993–995. [Google Scholar] [PubMed]
- Samuel, C.E. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 1993, 268, 7603–7606. [Google Scholar] [PubMed]
- Hershey, J.W. Translational control in mammalian cells. Annu. Rev. Biochem 1991, 60, 717–755. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Galabru, J. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur. J. Biochem. 1987, 167, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef] [PubMed]
- Daher, A.; Laraki, G.; Singh, M.; Melendez-Pena, C.E.; Bannwarth, S.; Peters, A.H.; Meurs, E.F.; Braun, R.E.; Patel, R.C.; Gatignol, A. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol. Cell. Biol. 2009, 29, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: The 2'-5'oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. N.Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 2008, 29, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, E.; Coch, C.; Wimmenauer, V.; Janke, M.; Barchet, W.; Hartmann, G. Approaching the RNA ligand for RIG-I? Immunol. Rev. 2009, 227, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Kerr, I.M. The (2'-5') oligoadenylate (pppA2'-5'A2'-5'A) synthetase and protein kinase(s) from interferon-treated cells. Eur. J. Biochem. 1979, 93, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Manche, L.; Green, S.R.; Schmedt, C.; Mathews, M.B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 1992, 12, 5238–5248. [Google Scholar] [PubMed]
- Michelson, A.M.; Massoulie, J.; Guschlbauer, W. Synthetic polynucleotides. Progr. Nucl. Acid. Res. Mol.Biol. 1967, 6, 83–141. [Google Scholar] [CrossRef]
- Schlee, M.; Barchet, W.; Hornung, V.; Hartmann, G. Beyond double-stranded RNA-type I IFN induction by 3pRNA and other viral nucleic acids. Curr. Top. Microbiol. Immunol. 2007, 316, 207–230. [Google Scholar] [PubMed]
- Nallagatla, S.R.; Hwang, J.; Toroney, R.; Zheng, X.; Cameron, C.E.; Bevilacqua, P.C. 5'-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 2007, 318, 1455–1458. [Google Scholar] [CrossRef] [PubMed]
- Nallagatla, S.R.; Bevilacqua, P.C. Nucleoside modifications modulate activation of the protein kinase PKR in an RNA structure-specific manner. Rna 2008, 14, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. 2006, 314, 997–1001. [Google Scholar] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; Endres, S.; Hartmann, G. 5'-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Myong, S.; Cui, S.; Cornish, P.V.; Kirchhofer, A.; Gack, M.U.; Jung, J.U.; Hopfner, K.P.; Ha, T. Cytosolic viral sensor RIG-I is a 5'-triphosphate-dependent translocase on double-stranded RNA. Science 2009, 323, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; Juranek, S.; Kato, H.; Kawai, T.; Poeck, H.; Fitzgerald, K.A.; Takeuchi, O.; Akira, S.; Tuschl, T.; Latz, E.; Ludwig, J.; Hartmann, G. Recognition of 5' triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Schwerd, T.; Hamm, W.; Hellmuth, J. C.; Cui, S.; Wenzel, M.; Hoffmann, F.S.; Michallet, M.C.; Besch, R.; Hopfner, K.P.; Endres, S.; Rothenfusser, S. 5'-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA 2009, 106, 12067–12072. [Google Scholar] [CrossRef]
- Cazenave, C.; Uhlenbeck, O.C. RNA template-directed RNA synthesis by T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 1994, 91, 6972–6976. [Google Scholar] [CrossRef]
- Nacheva, G.A.; Berzal-Herranz, A. Preventing nondesired RNA-primed RNA extension catalyzed by T7 RNA polymerase. Eur. J. Biochem. 2003, 270, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Schonborn, J.; Oberstrass, J.; Breyel, E.; Tittgen, J.; Schumacher, J.; Lukacs, N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991, 19, 2993–3000. [Google Scholar] [CrossRef] [PubMed]
- Colby, C.; Jurale, C.; Kates, J.R. Mechanism of synthesis of vaccinia virus double-stranded ribonucleic acid in vivo and in vitro . J. Virol. 1971, 7, 71–76. [Google Scholar] [PubMed]
- Jacquemont, B.; Roizman, B. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 1975, 15, 707–713. [Google Scholar]
- Pettersson, U.; Philipson, L. Synthesis of complementary RNA sequences during productive adenovirus infection. Proc. Natl. Acad. Sci. USA 1974, 71, 4887–4891. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Budt, M.; Niederstadt, L.; Valchanova, R.S.; Jonjic, S.; Brune, W. Specific inhibition of the PKR-mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J. Virol. 2009, 83, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, S.C.; Schur, P.H. Immunofluorescent localization of double-stranded RNA in reovirus-infected cells. Virology 1970, 41, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Marshall, J.A.; Bowden, D.S. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 1994, 200, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Stollar, B.D.; Stollar, V. Immunofluorescent demonstration of double-stranded RNA in the cytoplasm of Sindbis virus-infected cells. Virology 1970, 42, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Lavrova, I.K.; Voroshilova, M.K.; Chumakov, M.P.; Poverenny, A.M.; Podgorodnichenko, V.K. Immunofluorescent demonstration of double-stranded RNA and virus antigen in RNA virus-infected cells. Virology 1974, 62, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Boulant, S.; McLauchlan, J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008, 82, 2182–2195. [Google Scholar] [CrossRef] [PubMed]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [PubMed]
- Jacobs, B.L.; Langland, J.O. When two strands are better than one: The mediators and modulators of the cellular responses to double-stranded RNA. Virology 1996, 219, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Vyas, J.; Elia, A.; Clemens, M.J. Inhibition of the protein kinase PKR by the internal ribosome entry site of hepatitis C virus genomic RNA. RNA 2003, 9, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Shimoike, T.; McKenna, S.A.; Lindhout, D.A.; Puglisi, J.D. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antiviral Res. 2009, 83, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Carpick, B.W.; Graziano, V.; Schneider, D.; Maitra, R.K.; Lee, X.; Williams, B.R. Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J. Biol. Chem. 1997, 272, 9510–9516. [Google Scholar] [CrossRef] [PubMed]
- Edery, I.; Petryshyn, R.; Sonenberg, N. Activation of double-stranded RNA-dependent kinase (dsl) by the TAR region of HIV-1 mRNA: A novel translational control mechanism. Cell 1989, 56, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.K.; McMillan, N.A.; Desai, S.; McSwiggen, J.; Hovanessian, A.G.; Sen, G.; Williams, B.R.; Silverman, R.H. HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology 1994, 204, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Martinez-Sobrido, L.; Schneider, J.; Hai, R.; Waibler, Z.; Kalinke, U.; Garcia-Sastre, A.; Wolff, T. Influenza B virus ribonucleoprotein is a potent activator of the antiviral kinase PKR . PLoS Pathog. 2009, 5, e1000473. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, L.A.; Wong, C.J.; Lary, J.; Nallagatla, S.R.; Diegelman-Parente, A.; Zheng, X.; Cole, J.L.; Bevilacqua, P.C. RNA dimerization promotes PKR dimerization and activation. J. Mol. Biol. 2009, 390, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Ben-Asouli, Y.; Banai, Y.; Pel-Or, Y.; Shir, A.; Kaempfer, R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell 2002, 108, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Watson, J.C. In vitro activation of the interferon-induced, double-stranded RNA-dependent protein kinase PKR by RNA from the 3' untranslated regions of human alpha-tropomyosin. Proc. Natl. Acad. Sci. USA 1996, 93, 508–513. [Google Scholar] [CrossRef]
- Majde, J.A. Viral double-stranded RNA, cytokines, and the flu. J. Interferon Cytokine Res. 2000, 20, 259–272. [Google Scholar] [PubMed]
- Palese, P.; Shaw, M. Orthomyxoviridae: The viruses and their replication. In Fields - Virology, 5th edKnipe, D.M., Howley, P.M., Eds.; 2007; Lippincott Williams & Wilkins: Philadelphia, PA, USA. [Google Scholar]
- Tian, B.; Mathews, M.B. Functional characterization of and cooperation between the double-stranded RNA-binding motifs of the protein kinase PKR. J. Biol. Chem. 2001, 276, 9936–9944. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Hartmann, R.; Qin, J.; Sen, G.C. Modular structure of PACT: Distinct domains for binding and activating PKR. Mol. Cell. Biol. 2001, 21, 1908–1920. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.T.; White, C.L.; Peters, G.A.; Williams, B.R.; Sen, G.C. The role of PACT in mediating gene induction, PKR activation, and apoptosis in response to diverse stimuli. J. Interferon Cytokine Res. 2008, 28, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Schneider, J.; Wolff, T. Double-stranded RNA binding of influenza B virus nonstructural NS1 protein inhibits protein kinase R but is not essential to antagonize production of alpha/beta interferon. J. Virol. 2006, 80, 11667–11677. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.; Saito, S.; Fukuda, R. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J. Virol. 1999, 73, 2425–2433. [Google Scholar] [PubMed]
- Dauber, B.; Heins, G.; Wolff, T. The influenza B virus nonstructural NS1 protein is essential for efficient viral growth and antagonizes beta interferon induction. J. Virol. 2004, 78, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Donelan, N.R.; Dauber, B.; Wang, X.; Basler, C.F.; Wolff, T.; Garcia-Sastre, A. The N- and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 2004, 78, 11574–11582. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Wang, X.; Ehrhardt, C.; Zheng, H.; Donelan, N.; Planz, O.; Pleschka, S.; Garcia-Sastre, A.; Heins, G.; Wolff, T. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J. Virol. 2002, 76, 11166–11171. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale Jr., M.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus . J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Riedel, K.; Lynch, P.; Chien, C.Y.; Montelione, G.T.; Krug, R.M. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA 1999, 5, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Cheong, H.K.; Lee, J.H.; Cheong, C.; Kainosho, M.; Choi, B.S. Structural features of an influenza virus promoter and their implications for viral RNA synthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10602–10607. [Google Scholar] [CrossRef]
- Hsu, M.T.; Parvin, J.D.; Gupta, S.; Krystal, M.; Palese, P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc. Natl. Acad. Sci. USA 1987, 84, 8140–8144. [Google Scholar] [CrossRef]
- Klumpp, K.; Ruigrok, R.W.; Baudin, F. Roles of the influenza virus polymerase and nucleoprotein in forming a functional RNP structure. EMBO J. 1997, 16, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, C.; Nichol, S. Bunyaviridae: The viruses and their replication. In Fields - Virology, Knipe, D.M., Howley, P.M., Eds. 2007; Lippincott Williams & Wilkins: Philadelphia, PA, USA. [Google Scholar]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Overby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation . PLoS Pathog. 2009, 5, e1000287. [Google Scholar] [CrossRef] [PubMed]
- Streitenfeld, H.; Boyd, A.; Fazakerley, J.K.; Bridgen, A.; Elliott, R.M.; Weber, F. Activation of PKR by Bunyamwera virus is independent of the viral interferon antagonist NSs. J. Virol. 2003, 77, 5507–5511. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Cerveny, M.; Yan, Z.; He, B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007, 81, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Schumann, M.; Gantke, T.; Muhlberger, E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J. Virol. 2009, 83, 8993–8997. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Abraham, N.; Knowles, S.; Marius, R.; Brasey, A.; Lichty, B.D.; Brown, E.G.; Sonenberg, N.; Bell, J.C. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000, 74, 9580–9585. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; Mirazimi, A.; Weber, F. Processing of genome 5' termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction . PloS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Coloma, R.; Valpuesta, J.M.; Arranz, R.; Carrascosa, J.L.; Ortin, J.; Martin-Benito, J. The structure of a biologically active influenza virus ribonucleoprotein complex . PLoS Pathog. 2009, 5, e1000491. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.; Wernimont, A.K.; Muziol, T.; Ravelli, R.B.; Clapier, C.R.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 2006, 313, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Zhang, X.; Wertz, G.W.; Luo, M. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 2006, 313, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Leppert, M.; Rittenhouse, L.; Perrault, J.; Summers, D.F.; Kolakofsky, D. Plus and minus strand leader RNAs in negative strand virus-infected cells. Cell 1979, 18, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Kiley, M.P.; Holloway, B.P.; Auperin, D.D. Sequence analysis of the Ebola virus genome: Organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 1993, 29, 215–240. [Google Scholar] [CrossRef] [PubMed]
- Plumet, S.; Herschke, F.; Bourhis, J.M.; Valentin, H.; Longhi, S.; Gerlier, D. Cytosolic 5'-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response . PloS ONE 2007, 2, e279. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Musiyenko, A.; Bayfield, M.A.; Maraia, R.J.; Barik, S. Cellular La protein shields nonsegmented negative-strand RNA viral leader RNA from RIG-I and enhances virus growth by diverse mechanisms. J. Virol. 2008, 82, 7977–7987. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Komatsu, T.; Kitagawa, Y.; Sada, K.; Gotoh, B. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J. Virol. 2008, 82, 10102–10110. [Google Scholar] [CrossRef] [PubMed]
- Toth, A. M.; Devaux, P.; Cattaneo, R.; Samuel, C.E. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J. Virol. 2009, 83, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Reutter, G.L.; Cortese-Grogan, C.; Wilson, J.; Moyer, S.A. Mutations in the measles virus C protein that up regulate viral RNA synthesis. Virology 2001, 285, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Gainey, M.D.; Dillon, P.J.; Clark, K.M.; Manuse, M.J.; Parks, G.D. Paramyxovirus-induced shutoff of host and viral protein synthesis: Role of the P and V proteins in limiting PKR activation. J. Virol. 2008, 82, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Gale Jr., M.; Katze, M.G. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase . Pharmacol. Ther. 1998, 78, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Langland, J.O.; Cameron, J. M.; Heck, M.C.; Jancovich, J.K.; Jacobs, B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006, 119, 100–110. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Li, Z.; Okonski, K.M.; Toth, A.M.; Wang, Y.; Samuel, C.E. Tipping the balance: Antagonism of PKR kinase and ADAR1 deaminase functions by virus gene products. J. Interferon Cytokine Res. 2009, 29, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- de la Luna, S.; Fortes, P.; Beloso, A.; Ortin, J. Influenza virus NS1 protein enhances the rate of translation initiation of viral mRNAs. J. Virol. 1995, 69, 2427–2433. [Google Scholar] [PubMed]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Fortes, P.; Lamond, A.I.; Ortin, J. Influenza virus NS1 protein alters the subnuclear localization of cellular splicing components. J. Gen. Virol. 1995, 76, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qian, X.Y.; Krug, R.M. The influenza virus NS1 protein: A novel inhibitor of pre-mRNA splicing. Genes Dev. 1994, 8, 1817–1828. [Google Scholar] [CrossRef]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2'-5' oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef]
- Park, Y.W.; Katze, M.G. Translational control by influenza virus. Identification of cis-acting sequences and trans-acting factors which may regulate selective viral mRNA translation. J. Biol. Chem. 1995, 270, 28433–28439. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Krug, R.M. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol. 1994, 68, 2425–2432. [Google Scholar] [PubMed]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Dauber, B.; Melen, K.; Julkunen, I.; Wolff, T. Analysis of influenza B Virus NS1 protein trafficking reveals a novel interaction with nuclear speckle domains. J. Virol. 2009, 83, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Khan, J.A.; Swapna, G.V.; Ertekin, A.; Krug, R.M.; Tong, L.; Montelione, G.T. Conserved surface features form the double-stranded RNA binding site of non-structural protein 1 (NS1) from influenza A and B viruses. J. Biol. Chem. 2007, 282, 20584–20592. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Krug, R.M. The RNA-binding and effector domains of the viral NS1 protein are conserved to different extents among influenza A and B viruses. Virology 1996, 223, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.; Fukuda, R. Binding of influenza A virus NS1 protein to dsRNA in vitro . J. Gen. Virol. 1992, 73, 3325–3329. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Katze, M.G. Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J. Interferon Cytokine Res. 1998, 18, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.G.; Tomita, J.; Hovanessian, A.G.; Katze, M.G. Characterization and regulation of the 58,000-dalton cellular inhibitor of the interferon-induced, dsRNA-activated protein kinase. J. Biol. Chem. 1992, 267, 14238–14243. [Google Scholar] [PubMed]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 2007, 81, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.G.; Fornek, J.L.; Medigeshi, G.R.; Perrone, L.A.; Peng, X.; Dyer, M.D.; Proll, S.C.; Knoblaugh, S.E.; Carter, V.S.; Korth, M.J.; Nelson, J.A.; Tumpey, T.M.; Katze, M.G. P58(IPK): A novel "CIHD" member of the host innate defense response against pathogenic virus infection . PLoS Pathog. 2009, 5, e1000438. [Google Scholar] [CrossRef] [PubMed]
- Billecocq, A.; Spiegel, M.; Vialat, P.; Kohl, A.; Weber, F.; Bouloy, M.; Haller, O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004, 78, 9798–9806. [Google Scholar] [CrossRef] [PubMed]
- Blakqori, G.; Delhaye, S.; Habjan, M.; Blair, C.D.; Sanchez-Vargas, I.; Olson, K.E.; Attarzadeh-Yazdi, G.; Fragkoudis, R.; Kohl, A.; Kalinke, U.; Weiss, S.; Michiels, T.; Staeheli, P.; Weber, F. La Crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J. Virol. 2007, 81, 4991–4999. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Narayanan, K.; Worthy, M.; Peters, C.J. The S segment of Punta Toro virus (Bunyaviridae, Phlebovirus) is a major determinant of lethality in the Syrian hamster and codes for a type I interferon antagonist. J. Virol. 2007, 81, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Blakqori, G.; Wagner, V.; Banholzer, M.; Kessler, N.; Elliott, R. M.; Haller, O.; Weber, F. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J. Biol. Chem. 2004, 279, 31471–31477. [Google Scholar] [CrossRef] [PubMed]
- Black, T.L.; Safer, B.; Hovanessian, A.; Katze, M.G. The cellular 68,000-Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: Implications for translational regulation. J. Virol. 1989, 63, 2244–2251. [Google Scholar] [PubMed]
- Black, T.L.; Barber, G.N.; Katze, M.G. Degradation of the interferon-induced 68,000-M(r) protein kinase by poliovirus requires RNA. J. Virol. 1993, 67, 791–800. [Google Scholar] [PubMed]
- Basler, C.F.; Mikulasova, A.; Martinez-Sobrido, L.; Paragas, J.; Muhlberger, E.; Bray, M.; Klenk, H.D.; Palese, P.; Garcia-Sastre, A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003, 77, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Wang, X.; Muhlberger, E.; Volchkov, V.; Paragas, J.; Klenk, H.D.; Garcia-Sastre, A.; Palese, P. The Ebola virus VP35 protein functions as a type I IFN antagonist . Proc. Natl. Acad. Sci. USA 2000, 97, 12289–12294. [Google Scholar] [CrossRef]
- Chang, T.H.; Kubota, T.; Matsuoka, M.; Jones, S.; Bradfute, S.B.; Bray, M.; Ozato, K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery . PLoS Pathog. 2009, 5, e1000493. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Cardenas, W.B.; Basler, C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, L.; Sun, Y.; Nabel, G.J. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell. 2002, 10, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Muhlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.D.; Becker, S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 1999, 73, 2333–2342. [Google Scholar] [PubMed]
- Reid, S.P.; Cardenas, W.B.; Basler, C.F. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology 2005, 341, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, W.B.; Loo, Y.M.; Gale Jr., M.; Hartman, A.L.; Kimberlin, C.R.; Martinez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling . J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 2004, 328, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Bird, B.H.; Towner, J.S.; Antoniadou, Z.A.; Zaki, S.R.; Nichol, S.T. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J. Virol. 2008, 82, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Ling, L.; Nichol, S.T.; Hibberd, M.L. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008, 82, 5348–5358. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Hakki, M.; de Niro, K.L.; Geballe, A.P. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 2004, 78, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Geballe, A.P. Double-stranded RNA binding by human cytomegalovirus pTRS1. J. Virol. 2005, 79, 7311–7318. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, C.A.; Shenk, T. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J. Virol. 2002, 76, 12290–12299. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.R.; Muzithras, V.P. A cluster of dispensable genes within the human cytomegalovirus genome short component: IRS1, US1 through US5, and the US6 family. J. Virol. 1992, 66, 2541–2546. [Google Scholar] [PubMed]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A.; Kouzarides, T.; Martignetti, J.A.; Preddie, E.; Satchwell, S.C.; Tomlinson, P.; Weston, K.M.; Barrell, B.G. nalysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169 . A. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar]
- Menard, C.; Wagner, M.; Ruzsics, Z.; Holak, K.; Brune, W.; Campbell, A.E.; Koszinowski, U.H. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 2003, 77, 5557–5570. [Google Scholar] [CrossRef] [PubMed]
- Valchanova, R.S.; Picard-Maureau, M.; Budt, M.; Brune, W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J. Virol. 2006, 80, 10181–10190. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Geballe, A.P. Binding and relocalization of protein kinase R by murine cytomegalovirus. J. Virol. 2009, 83, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Hanson, L.K.; Brown, C.E.; Janzen, D.M.; Geballe, A.P. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J. Virol. 2006, 80, 10173–10180. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Rothenburg, S.; Seo, E.J.; Gibbs, J.S.; Dever, T.E.; Dittmar, K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 2009, 16, 63–70. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Dauber, B.; Wolff, T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses 2009, 1, 523-544. https://doi.org/10.3390/v1030523
Dauber B, Wolff T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses. 2009; 1(3):523-544. https://doi.org/10.3390/v1030523
Chicago/Turabian StyleDauber, Bianca, and Thorsten Wolff. 2009. "Activation of the Antiviral Kinase PKR and Viral Countermeasures" Viruses 1, no. 3: 523-544. https://doi.org/10.3390/v1030523
APA StyleDauber, B., & Wolff, T. (2009). Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses, 1(3), 523-544. https://doi.org/10.3390/v1030523