
Metabolically Active Prokaryotic Complex in Grassland and Forests’ Sod-Podzol under Polycyclic Aromatic Hydrocarbon Influence

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
Research Methods
3. Results
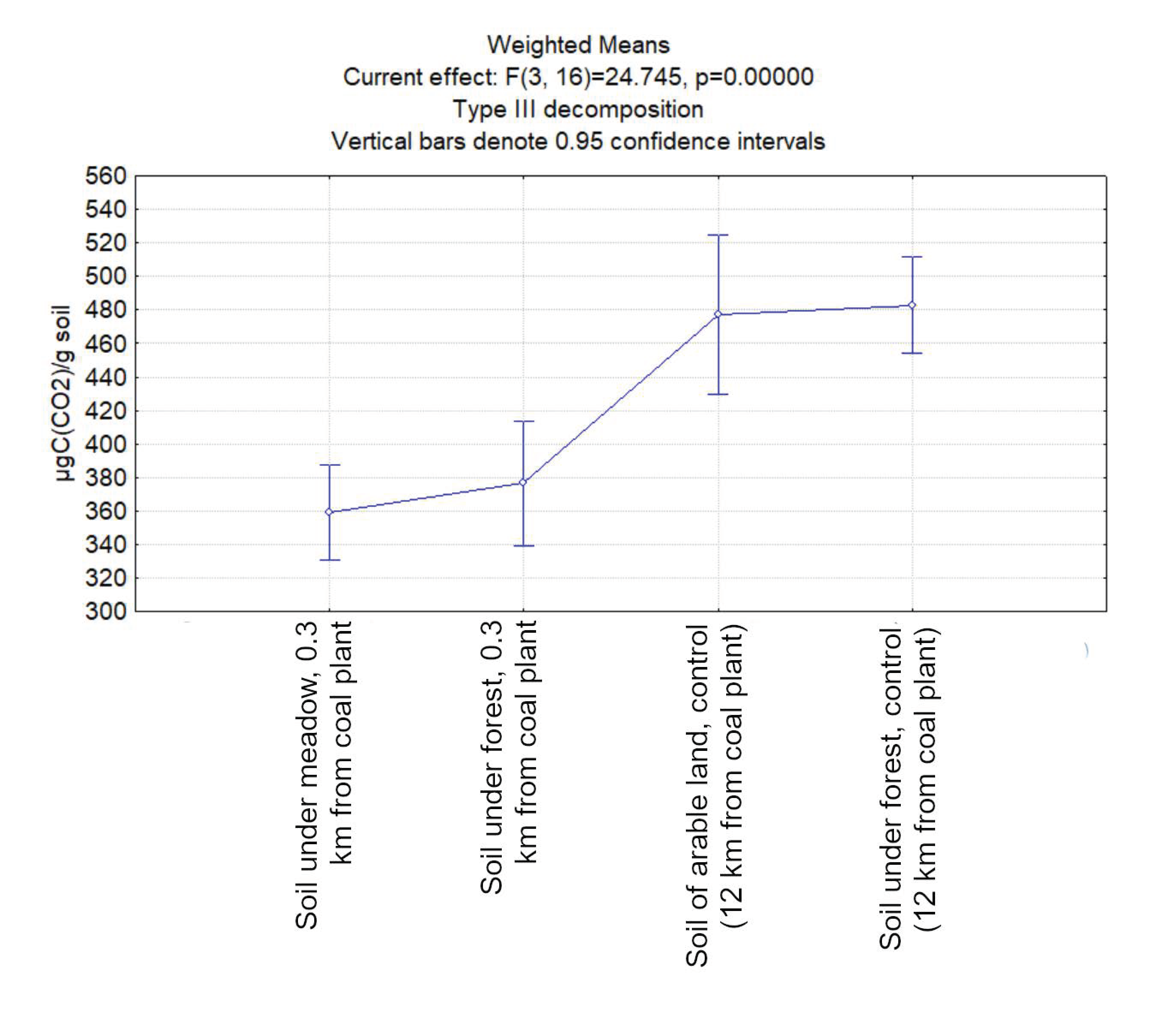
3.1. Carbon Dioxide Emission from the Studied Soil Samples
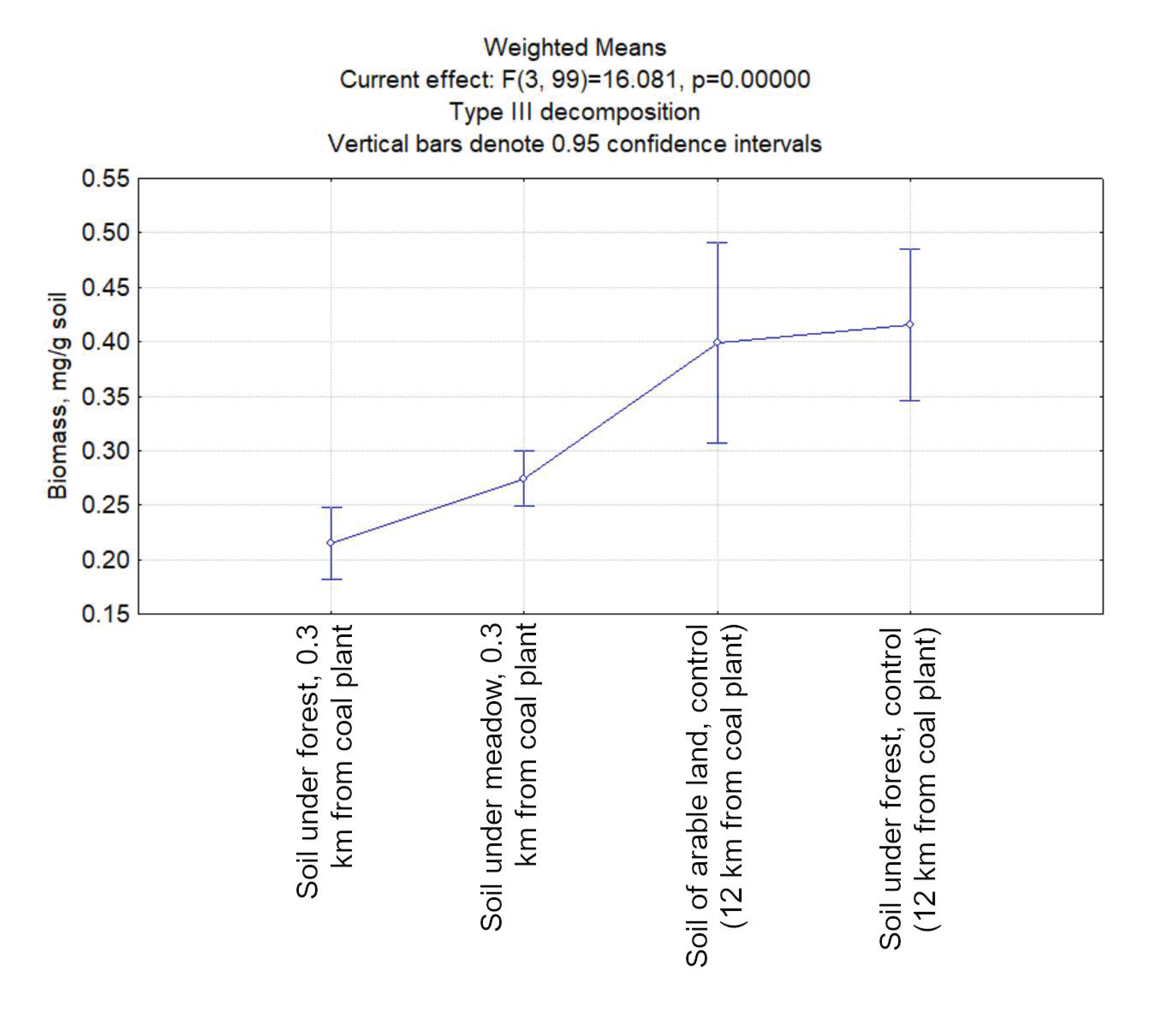
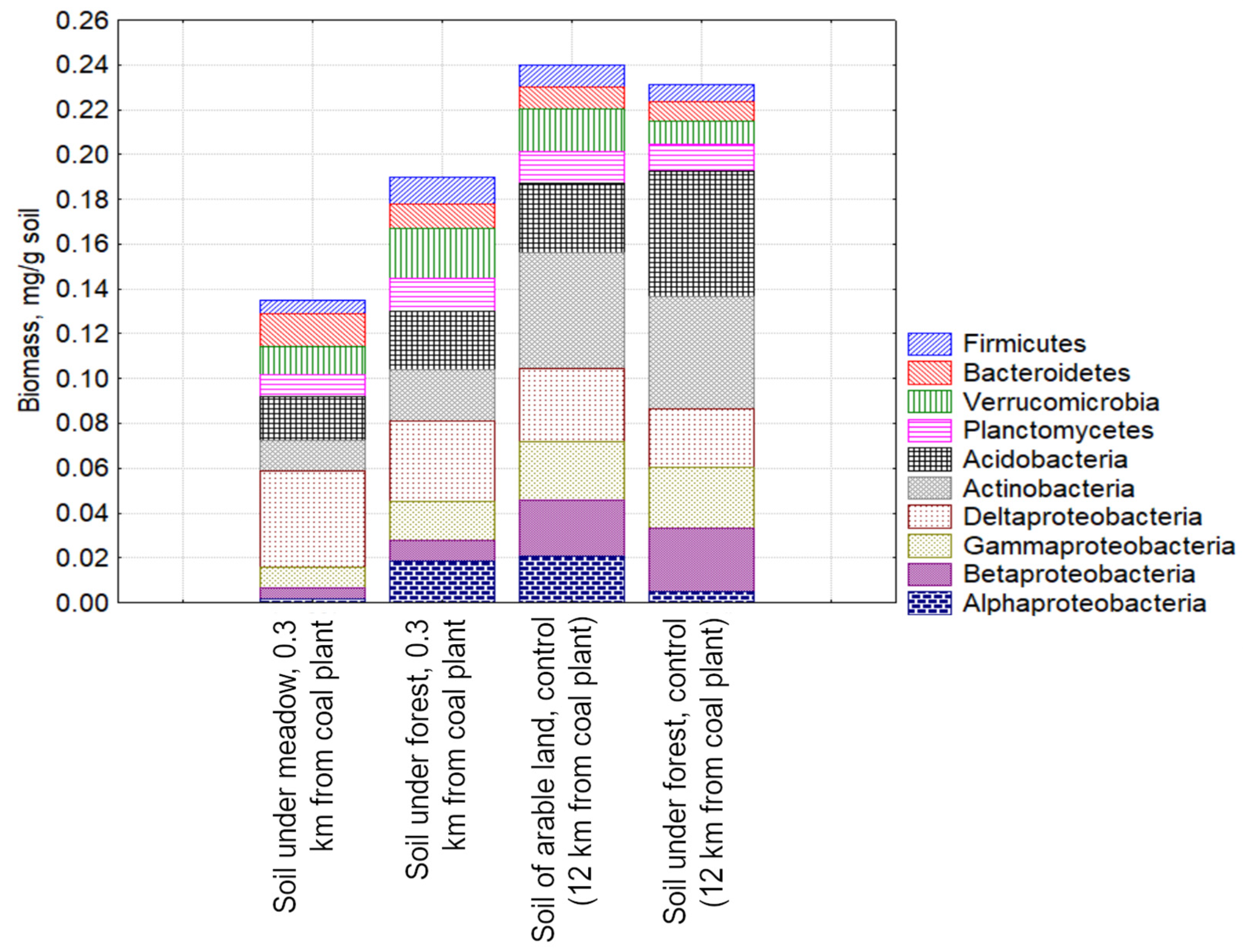
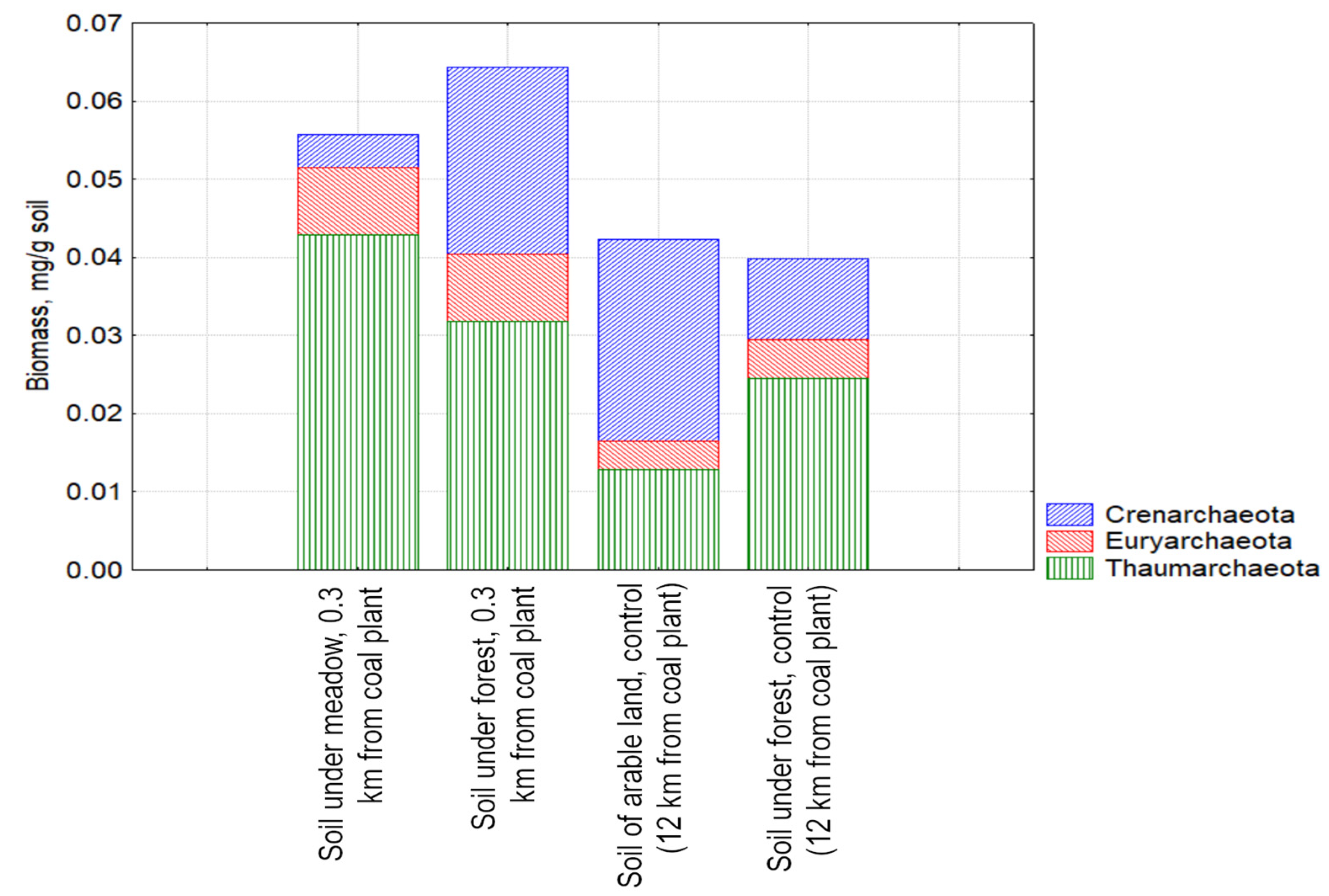
3.2. The Biomass of Prokaryotes in the Studied Soil Samples
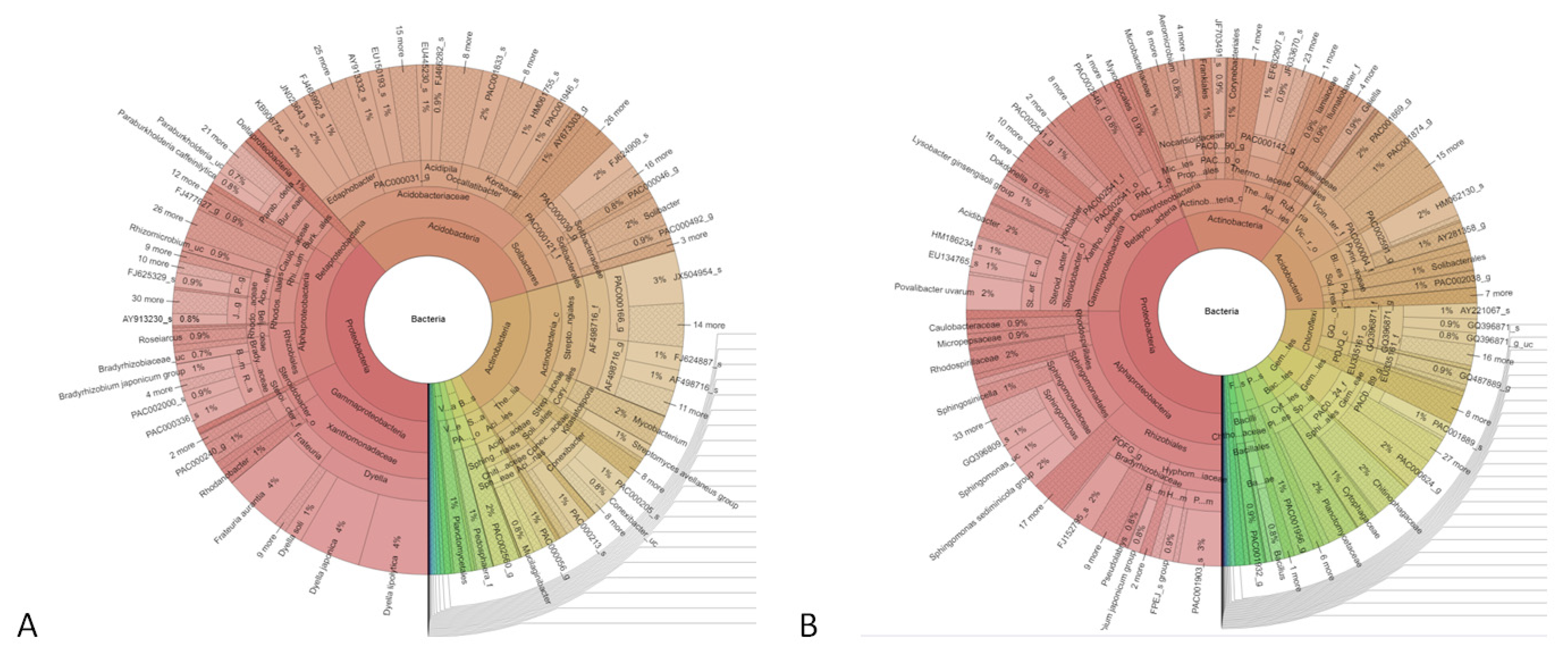
3.3. Study of the Structure of the Metabolically Active Prokaryotic Complex
3.4. Detection of the Presence of the Functional Alkane Monooxygenase (alkB) Geneby RT-PCR and Both the Abundance and Biomass of Metabolically Active Prokaryotes Carrying Functional Alkane Monooxygenase by FISH
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gennadiev, A.N.; Pikovskii, Y.I.; Zhidkin, A.P.; Kovach, R.G.; Koshovskii, T.S.; Smirnova, M.A.; Khlynina, N.I.; Tsibart, A.S. Factors and features of the hydrocarbon status of soils. Eurasian Soil Sci. 2015, 48, 1193–1206. [Google Scholar] [CrossRef]
- Nazina, T.N.; Sokolova, D.S.; Babich, T.L.; Semenova, E.M.; Ershov, A.P.; Bidzhieva, S.K.; Borzenkov, I.A.; Tourova, T.P.; Poltaraus, A.B.; Khisametdinov, M.R. Microorganisms of low-temperature heavy oil reservoirs (Russia) and their possible application for enhanced oil recovery. Microbiology 2017, 86, 773–785. [Google Scholar] [CrossRef]
- Ivshina, I.B.; Kostina, L.V.; Kamenskikh, T.N.; Zhuikova, V.A.; Zhuikova, T.V.; Bezel, V.S. Soil microbiocenosis as an indicator of stability of meadow communities in the environment polluted with heavy metals. Russ. J. Ecol. 2014, 45, 83–89. [Google Scholar] [CrossRef]
- IUSS Working Group WRB. World Reference Base for Soil Resources 2014: International Soil Classification System for Naming Soils and Creating Legends for Soil Maps; World Soil Resources Reports; FAO: Rome, Italy, 2014; Volume 106, pp. 212–213. [Google Scholar]
- Gennadiev, A.N.; Zhidkin, A.P.; Koshovskii, T.S.; Lobanov, A.A. Polyarenes and bitumoids from analogous technogenic hydrocarbon sources with different parameters in soils. Eurasian Soil Sci. 2018, 51, 1369–1380. [Google Scholar] [CrossRef]
- Wilcke, W. Polycyclic aromatic hydrocarbons (PAHs) in soil, A review. J. Plant Nutr. Soil Sci. 2000, 163, 229–248. [Google Scholar] [CrossRef]
- Manucharova, N.A.; Ksenofontova, N.A.; Karimov, T.D.; Vlasova, A.P.; Zenova, G.M.; Stepanov, A.L. Changes in the phylogenetic structure of the metabolically active prokaryotic soil complex induced by oil pollution. Microbiology 2020, 89, 219–230. [Google Scholar] [CrossRef]
- Whyte, L.G.; Schultz, A.; van Beiden, J.B.; Luz, A.P.; Pellizari, V.; Labbé, D.; Greer, C.W. Prevalence of alkane monooxygenase genes in Arctic and Antarctic hydrocarbon-contaminated and pristine soils. FEMS Microbiol. Ecol. 2002, 41, 141–150. [Google Scholar] [CrossRef]
- Amann, R.I.; Krunholz, L.; Stahl, D.A. Fluorescent oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 1990, 172, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.A.; Amann, R. Development and application of nucleic acid probes in bacterial systematics. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons Ltd.: New York, NY, USA, 1991; pp. 205–248. [Google Scholar]
- Raskin, L.; Stromley, J.; Rittmann, B.; Stahl, D. Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl. Environ. Microbiol. 1994, 60, 1232–1240. [Google Scholar] [CrossRef]
- Teira, E.; Reinthaler, T.; Pernthaler, A.; Pernthaler, J.; Herndl, G.J. Combining catalyzed reporter deposition-fluorescence in situ hybridization and microautoradiography to detect substrate utilization by bacteria and archaea in the deep ocean. Appl. Environ. Microbiol. 2004, 70, 4411–4414. [Google Scholar] [CrossRef]
- Timonen, S.; Bomberg, M. Archaea in dry soil environments. Phytochem. Rev. 2009, 8, 505–518. [Google Scholar] [CrossRef]
- Manz, W.; Amann, R.; Ludwig, W.; Wagner, M.; Schleifer, K. Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria: Problems and solutions. Syst. Appl. Microbiol. 1992, 15, 593–600. [Google Scholar] [CrossRef]
- Dedysh, S.N.; Panikov, N.S.; Liesack, W.; Großkopf, R.; Zhou, J.; Tiedje, J.M. Isolation of acidophilic methane-oxidizing bacteria from northern peat wetlands. Science 1998, 282, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Rabus, R.; Wilkes, H.; Schramm, A.; Harms, G.; Behrends, A.; Amann, R.; Widdel, F. Anaerobic utilization of alkylbenzenes and n-alkanes from crude oil in an enrichment culture of denitrifying bacteria affiliating with the beta-subclass of Proteobacteria. Environ. Microbiol. 1999, 1, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Manz, W.; Amann, R.; Ludwig, W.; Vancanneyt, M.; Schleifer, K.-H. Application of a suite of 16S rRNA- specific oligonucleotide probes designed to investigate bacteria of the phylum cytophaga-flavobacter-bacteroides in the natural environment. Microbiology 1996, 142, 1097–1106. [Google Scholar] [CrossRef]
- Roller, C.; Wagner, M.; Amann, R.; Ludwig, W.; Schleifer, K.-H. In situ probing of Gram-positive bacteria with high DNA G + C content using 23s rRNA-targeted oligonucleotides. Microbiology 1995, 140, 2849–2858. [Google Scholar] [CrossRef]
- Meier, H.; Amann, R.; Ludwig, W.; Schleifer, K.H. Specific oligonucleotide probes for in situ detection of a major group of Gram-positive bacteria with low DNA G+C content. Syst. Appl. Microbiol. 1999, 22, 186–196. [Google Scholar] [CrossRef]
- Juretschko, S.; Loy, A.; Lehner, A.; Wagner, M. The microbial community composition of a nitrifying-denitrifying activated sludge from an industrial sewage treatment plant analyzed by the full-cycle rRNA approach. Syst. Appl. Microbiol. 2002, 25, 84–99. [Google Scholar] [CrossRef]
- Dedysh, S.N.; Pankratov, T.A.; Belova, S.E.; Kulichevskaya, I.S.; Liesack, W. Phylogenetic analysis and in situ identification of bacteria community composition in an acidic sphagnum peat bog. Appl. Environ. Microbiol. 2006, 72, 2110–2117. [Google Scholar] [CrossRef]
- Neef, A.; Amann, R.; Schlesner, H.; Schleifer, K.H. Monitoring a widespread bacterial group: In situ detection of planctomycetes with 16S rRNA-targeted probes. Microbiology 1998, 144, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Zhili, H.; Liyou, W.; Xingyuan, L.; Matthew, W.F.; Jizhong, Z. Empirical establishment of oligonucleotide probe design criteria. Appl. Environ. Microbiol. 2005, 71, 3753–3760. [Google Scholar] [CrossRef]
- Chen, Y. Application of Hydrocarbon Degrading Microorganism Enumeration and Catabolic Genes Detection for Soil Assessment. Master’s Thesis, University of Helsinki, Helsinki, Finland, September 2013. [Google Scholar]
- Manucharova, N.A.; Kuteinikova, Y.V.; Ivanov, P.V.; Nikolaeva, S.K.; Trofimov, V.T.; Stepanov, P.Y.; Tyapkina, E.V.; Lipatov, D.N.; Stepanov, A.L. Molecular analysis of the hydrolytic component of petroleum contaminated soils and of soils remediated with chitin. Microbiology 2017, 86, 395–402. [Google Scholar] [CrossRef]
- Kohno, T.; Sugimoto, Y.; Sei, K.; Mori, K. Design of PCR primers and gene probes for general detection of alkane-degrading bacteria. Microbes Environ. 2002, 17, 114–121. [Google Scholar] [CrossRef]
- Sei, K.; Sugimoto, Y.; Mori, K.; Maki, H.; Kohno, T. Monitoring of alkane-degrading bacteria in a sea-water microcosm during crude oil degradation by polymerase chain reaction based on alkanecatabolic genes. Environ. Microbiol. 2003, 5, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Moharikar, A.; Kapley, A.; Purohit, H.J. Detection of dioxygenase genes present in various activated sludge. Environ. Sci. Pollut. Res. Int. 2003, 10, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Teske, A.; Durbin, A.; Ziervogel, K.; Cox, C.; Arnosti, C. Microbial community composition and function in permanently cold seawater and sediments from an arctic fjord of Svalbard. Appl. Environ. Microbiol. 2011, 77, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.M.; Smits, T.H.; van Beilen, J.B.; Rojo, F. The alkane hydroxylase gene of Burkholderiacepacia RR10 is under catabolite repression control. J. Bacteriol. 2001, 183, 4202–4209. [Google Scholar] [CrossRef]
- Baek, K.H.; Yoon, B.D.; Kim, B.H.; Cho, D.H.; Lee, I.S.; Oh, H.M.; Kim, H.S. Monitoring of microbial diversity and activity during bioremediation of crude oil-contaminated soil with different treatments. J. Microbiol. Biotechnol. 2007, 17, 67–73. [Google Scholar]
- van Beilen, J.B.; Smits, T.H.M.; Roos, F.F.; Brunner, T.; Balada, S.B.; Röthlisberger, M.; Witholt, B. Identification of an amino acid position that determines the substrate range of integral membrane alkane hydroxylases. J. Bacteriol. 2005, 187, 85–91. [Google Scholar] [CrossRef]
- Korshunova, A.V.; Tourova, T.P.; Shestakova, N.M.; Mikhailova, E.M.; Poltaraus, A.B.; Nazina, T.N. Detection and transcription of n-alkane biodegradation genes (alkB) in the genome of a hydrocarbon-oxidizing bacterium Geobacillus subterraneus K. Microbiology 2011, 80, 682–691. [Google Scholar] [CrossRef]
- Shapiro, T.; Chekanov, K.; Alexandrova, A.; Dolnikova, G.; Ivanova, E.; Lobakova, E. Revealing of non-cultivable bacteria associated with the mycelium of fungi in the kerosene-degrading community isolated from the contaminated jet fuel. J. Fungi 2021, 7, 43. [Google Scholar] [CrossRef]
- Karlapudi, A.P.; Venkateswarulu, T.C.; Tammineedi, J.; Kanumuri, L.; Ravuru, B.K.; ramu Dirisala, V.; Kodali, V.P. Role of biosurfactants in bioremediation of oil pollution—A review. Petroleum 2018, 4, 241–249. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Soil | Sampling Location | Sampling Depth, cm |
|---|---|---|---|
| 1 | Sod-podzolic post-agrogenic podzolized clayey-illuviated | City, meadow, 0.3 km from the plant (55.728016, 38.217825) | 5–10 |
| 2 | Sod-podzolic peated artistratified clayey-illuviated | City, forest, 0.3 km from the plant (55.728355, 38.220142) | 5–10 |
| 3 | Sod-podzolic gley ultra-deeply lightened | Forest, 12 km from the plant (55.724166, 38.389851) | 5–10 |
| 4 | Sod-podzolic gley ultra-deeply lightened | Arable land, 12 km (55.724371, 38.387340) | 5–10 |
| Sample | Phenanthrene, ng/g Soil | Chrysene, ng/g Soil | Anthracene, ng/g Soil | Benz(a)pyrene, ng/g Soil | PAH content, ng/g of Soil | Corg, % |
|---|---|---|---|---|---|---|
| 1 | 19,289.5 | 1937.5 | 1033.0 | 514.6 | 26,818.1 | 4.6 |
| 2 | 9416.9 | 6344.0 | 677.1 | 1518.7 | 29,128.3 | 10.8 |
| 3 | 133.7 | 40.9 | 14.5 | 2.0 | 443.1 | 3.3 |
| 4 | 44.1 | 4.6 | 0.0 | 0.1 | 74.3 | 2.9 |
| Probe | Target Group of Organisms | 16S rRNA Target Site | Nucleotide Sequence of the Probe (5’-3’) | Formamide, % a | NaCl, mM b | Reference |
|---|---|---|---|---|---|---|
| EUB338 I | Bacteria | 338–355 | GCTGCC TCC | 20 | 225 | [9] |
| EUB338 II | Bacteria (Planctomycetales) | CGTAGGAGTGCAGCCACCC | ||||
| EUB338 III | Bacteria (Verrucomicrobiales) | GTAGGTGTGCTGCCACCCGTAGGTGT | ||||
| ARCH915 | Archaea | 915–934 | GTG CTC CCC CGC CAA TTC CT | 30 | 112 | [9,10] |
| ARC344 | 344–363 | TCGCGCCTGCTGCIC CCC GT | ||||
| EURY806 | Euryarchaeota | 806–824 | CAC AGC GTT TAC ACC TAG | 30 | 112 | [11,12] |
| CREN537 | Cren + Thaumarchaeota | 537–555 | TGA CCA CTT GAG GTG CTG | 30 | 112 | [11] |
| THAUM-494 | Thaumarchaeota | 494–513 | GAA TAA GGG GTG GGC AAGT | 30 | 112 | [11] |
| ALFlb | Alphaproteobacteria | 19–35 | CGTTCGYTCTGAGCCAG cG | 20 | 225 | [13,14] |
| ALF968 | 968–986 | GTAAGGTTCTGCGCGTT | ||||
| BET42a | Betaproteobacteria | 1027–1043 d | GCC TTC CCA CTT CGT TT | 35 | 80 | [13] |
| GAM42a | Gammaproteobacteria | 1027–1043 d | GCC TTC CCA CAT CGT TT | 35 | 80 | [13] |
| SRB385Db | Deltaproteobacteria | 385–402 | CGG CGT TGC TGC GTC AGG | 20 | 225 | [15] |
| CF319a | Bacteroidetes | 560–593 | TGG TCC GTG TCT CAG TAC WCC CTT | 35 | 80 | [16] |
| CFB560 | TAA ACC CART | |||||
| HGC69a | Actinobacteria | 1901–1918 d | TAT AGT TAC CAC CGC CGT e | 25 | 159 | [17] |
| LGC354A | Firmicutes | 354–371 | TGGGAAGATTCCCTACTGC | 35 | 80 | [18] |
| LGC354B | CGGGAAGATTCCCTACTGC | |||||
| LGC354C f | CCGGAAGATTCCCTACTGC | |||||
| HoAc1402 | Acidobacteria | 1402–1420 | CTT TCG TGA TGT GAC GGG | 10 | 450 | [19] |
| Ver138 | Verrucomicrobia | 138–155 | CGA GCT ATT CCC CTC TTG | 10 | 450 | [20] |
| Ver1455 | 1455–1472 | CCA TCC ATA CCT TCG GCA | ||||
| PLA46 | Planctomycetes | 46–64 | GAC TTG CAT GCC TAA TCC | 30 | 112 | [21,22] |
| PLA886 | 886–905 | GCC TTG CGA CCA TAC TCC C |
| Probe | Nucleotide Sequence of the Probe (5′-3′) | Formamide, % a | NaCl, mM b |
|---|---|---|---|
| alkB(F) | Cy3-tgg-ccg-gct-act-ccg-atg-atc-gga-atc-tgg | 50 | 500 |
| alkB(R) | Cy3-cgc-gtg-gtg-atc-cga-gtg-ccg-ctg-aag-gtg |
| Probe | Enzyme | Nucleotide Sequence of the Probe (5′-3′) | Size of the Target Area (bp) | Reference |
|---|---|---|---|---|
| alkB | alkanemonooxygenase | F TGGCCGGCTACTCCGATGATCGGAATCTGG | 870 | [8] |
| Re CGCGTGGTGATCCGAGTGCCGCTGAAGGTG |
| Soil | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Soil under forest vegetation, 0.3 km from coal plant | 0.361477 | 0.00000 | 0.000005 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.361477 | 0.000055 | 0.000031 | |
| Soil of arable land, control (12 km from coal plant) | 0.000009 | 0.000055 | 0.766284 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.000005 | 0.000031 | 0.766284 |
| Soil | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Soil under forest vegetation, 0.3 km from coal plant | 0.053527 | 0.000002 | 0.000000 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.053527 | 0.000868 | 0.000056 | |
| Soil of arable land, control (12 km from coal plant) | 0.000002 | 0.000868 | 0.676425 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.000000 | 0.000056 | 0.674225 |
| Soil | 1 | 2 | 3 | 4 | Mean | St. Err |
|---|---|---|---|---|---|---|
| Soil under forest vegetation, 0.3 km from coal plant | 0.2168 | 0.0039 | 0.0055 | 0.023 | 0.005 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.2168 | 0.0007 | 0.0009 | 0.014 | 0.003 | |
| Soil of arable land, control (12 km from coal plant) | 0.0039 | 0.0007 | 0.8177 | 0.052 | 0.007 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.0055 | 0.0009 | 0.8177 | 0.050 | 0.004 | |
| Soil under forest vegetation, 0.3 km from coal plant | 0.3639 | 0.5476 | 0.0033 | 0.026 | 0.003 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.3639 | 0.1504 | 0.0009 | 0.019 | 0.004 | |
| Soil of arable land, control (12 km from coal plant) | 0.5476 | 0.1504 | 0.0081 | 0.031 | 0.004 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.0033 | 0.0009 | 0.0081 | 0.056 | 0.008 | |
| Soil under forest vegetation, 0.3 km from coal plant | 0.4704 | 0.2454 | 0.0071 | 0.026 | 0.003 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.4704 | 0.6389 | 0.0201 | 0.019 | 0.004 | |
| Soil of arable land, control (12 km from coal plant) | 0.2454 | 0.6389 | 0.0396 | 0.031 | 0.004 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.0071 | 0.0201 | 0.0396 | 0.056 | 0.008 |
| Soil | 1 | 2 | 3 | 4 | Mean | St.Err |
|---|---|---|---|---|---|---|
| Soil under forest vegetation, 0.3 km from coal plant | 0.229421 | 0.055347 | 0.410960 | 0.031859 | 0.005774 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.229421 | 0.0007597 | 0.061951 | 0.042887 | 0.00866 | |
| Soil of arable land, control (12 km from coal plant) | 0.055347 | 0.007597 | 0.206866 | 0.012866 | 0.002887 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.410960 | 0.061951 | 0.206866 | 0.024507 | 0.005196 | |
| Soil under forest vegetation, 0.3 km from coal plant | 1.00000 | 0.000636 | 0.003636 | 0.008577 | 0.000751 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 1.00000 | 0.000636 | 0.003636 | 0.008577 | 0.000635 | |
| Soil of arable land, control (12 km from coal plant) | 0.000636 | 0.003636 | 0.212999 | 0.003676 | 0.000520 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.000636 | 0.003636 | 0.212999 | 0.004901 | 0.000635 | |
| Soil under forest vegetation, 0.3 km from coal plant | 0.002388 | 0.692936 | 0.016999 | 0.023894 | 0.005196 | |
| Soil under meadow vegetation, 0.3 km from coal plant | 0.002388 | 0.001396 | 0.0209428 | 0.004289 | 0.000577 | |
| Soil of arable land, control (12 km from coal plant) | 0.692936 | 0.001396 | 0.009192 | 0.025732 | 0.000981 | |
| Soil under forest vegetation, control (12 km from coal plant) | 0.016999 | 0.0209428 | 0.009192 | 0.010415 | 0.003464 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manucharova, N.A.; Pozdnyakov, L.A.; Vlasova, A.P.; Yanovich, A.S.; Ksenofontova, N.A.; Kovalenko, M.A.; Stepanov, P.Y.; Gennadiev, A.N.; Golovchenko, A.V.; Stepanov, A.L. Metabolically Active Prokaryotic Complex in Grassland and Forests’ Sod-Podzol under Polycyclic Aromatic Hydrocarbon Influence. Forests 2021, 12, 1103. https://doi.org/10.3390/f12081103
Manucharova NA, Pozdnyakov LA, Vlasova AP, Yanovich AS, Ksenofontova NA, Kovalenko MA, Stepanov PY, Gennadiev AN, Golovchenko AV, Stepanov AL. Metabolically Active Prokaryotic Complex in Grassland and Forests’ Sod-Podzol under Polycyclic Aromatic Hydrocarbon Influence. Forests. 2021; 12(8):1103. https://doi.org/10.3390/f12081103
Chicago/Turabian StyleManucharova, Natalia A., Lev A. Pozdnyakov, Anastasiya P. Vlasova, Anastasiya S. Yanovich, Natalia A. Ksenofontova, Maria A. Kovalenko, Pavel Y. Stepanov, Alexander N. Gennadiev, Alla V. Golovchenko, and Alexey L. Stepanov. 2021. "Metabolically Active Prokaryotic Complex in Grassland and Forests’ Sod-Podzol under Polycyclic Aromatic Hydrocarbon Influence" Forests 12, no. 8: 1103. https://doi.org/10.3390/f12081103
APA StyleManucharova, N. A., Pozdnyakov, L. A., Vlasova, A. P., Yanovich, A. S., Ksenofontova, N. A., Kovalenko, M. A., Stepanov, P. Y., Gennadiev, A. N., Golovchenko, A. V., & Stepanov, A. L. (2021). Metabolically Active Prokaryotic Complex in Grassland and Forests’ Sod-Podzol under Polycyclic Aromatic Hydrocarbon Influence. Forests, 12(8), 1103. https://doi.org/10.3390/f12081103