

Enhanced Viral Genome Classification Using Large Language Models

 ,
, 

Abstract
1. Introduction
2. Related Works
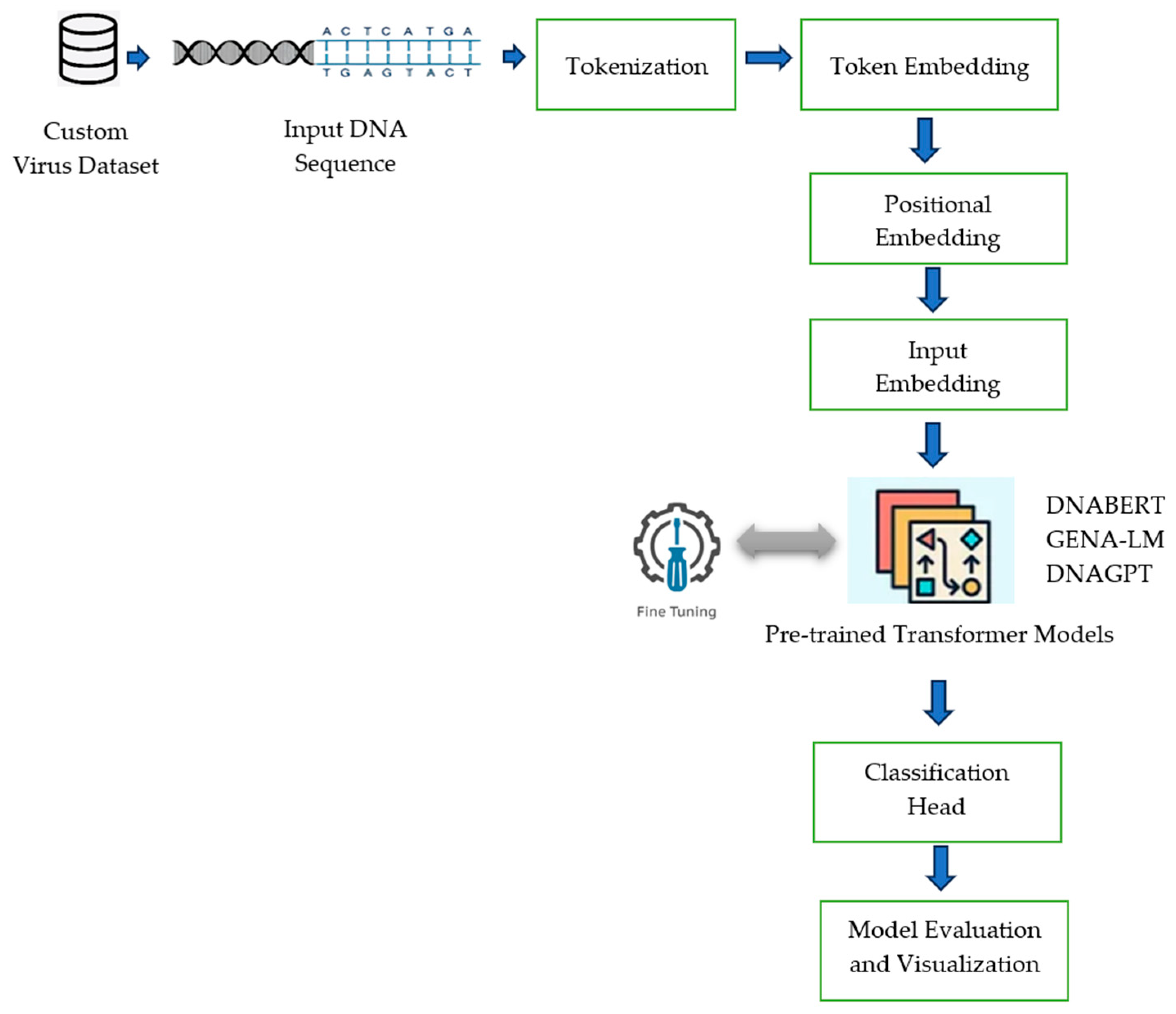
3. Methodology
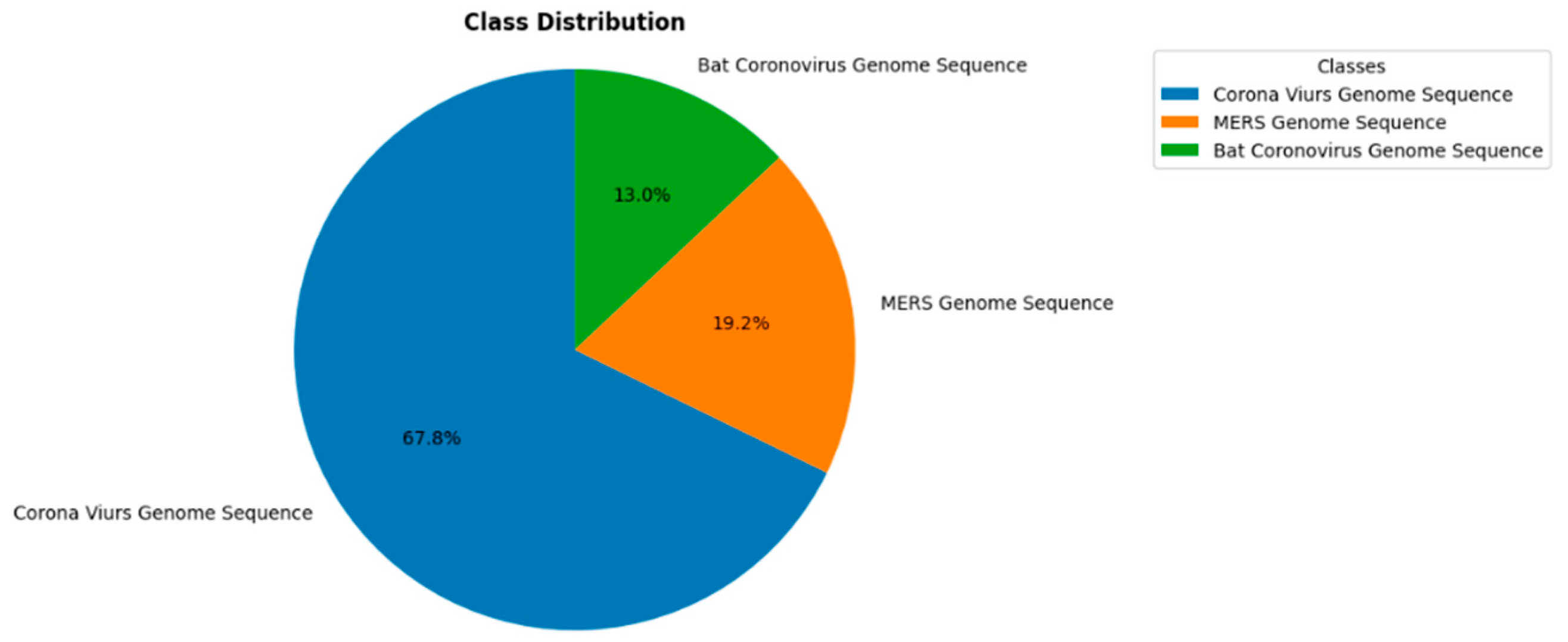
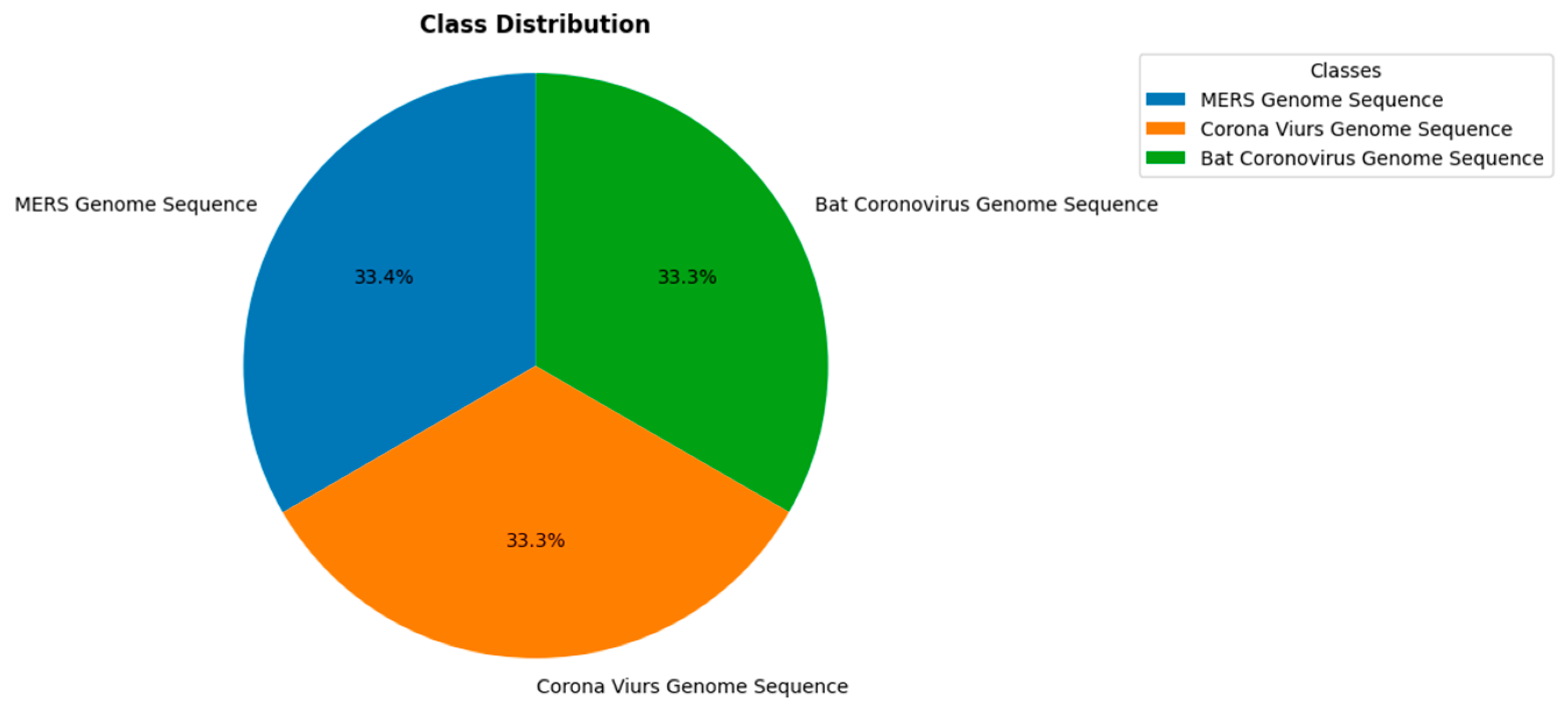
3.1. Dataset
3.2. Transformer
- The Attention Component;
- The Feedforward Component.
3.3. DNABERT
3.4. GENA-LM
3.5. DNAGPT
4. Materials and Methods
| Algorithm 1: Steps for DNA Sequence Classification |
| 1: Load the DNA sequence dataset 2: Download the pre-trained model and its respective tokenization for the models: DNAGPT, DNABERT-S, DNABERT-2, and GENL-LM from the Hugging Face library. Model <--- Load the model from the Hugging Face library 3: Apply the tokenizer to convert the input DNA sequence to a sequence of tokens. 4: Embedding: For each tokenized sequence, convert the tokens into dense vectors of size 768. 5: Add the Transformer layers and the task-specific (classification head) to the model. 6: Fine-tune the pre-trained model using a custom dataset to optimize its parameters for the specific classification task. 7: Measure the model’s performance using appropriate metrics (e.g., accuracy, F1-score). |
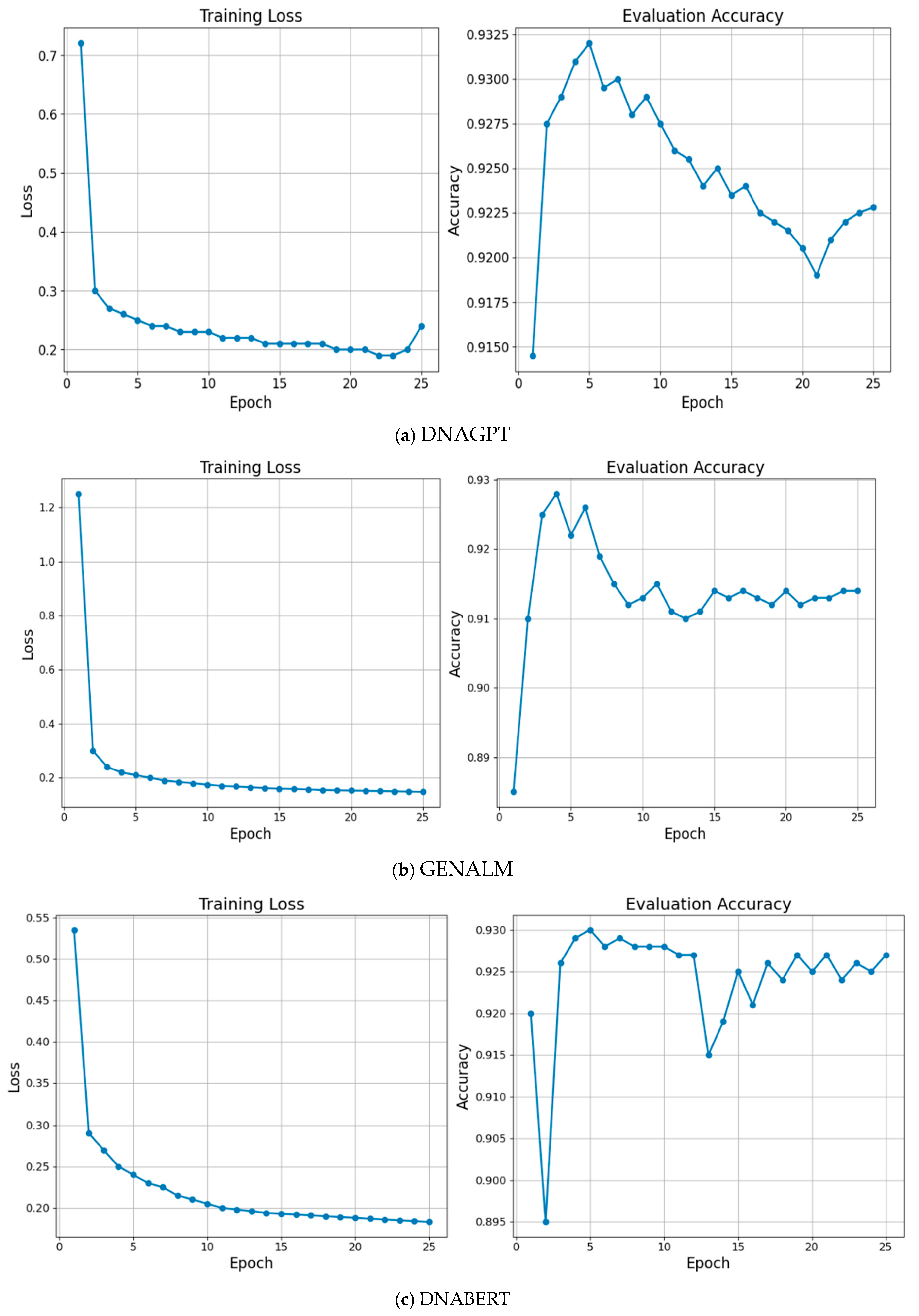
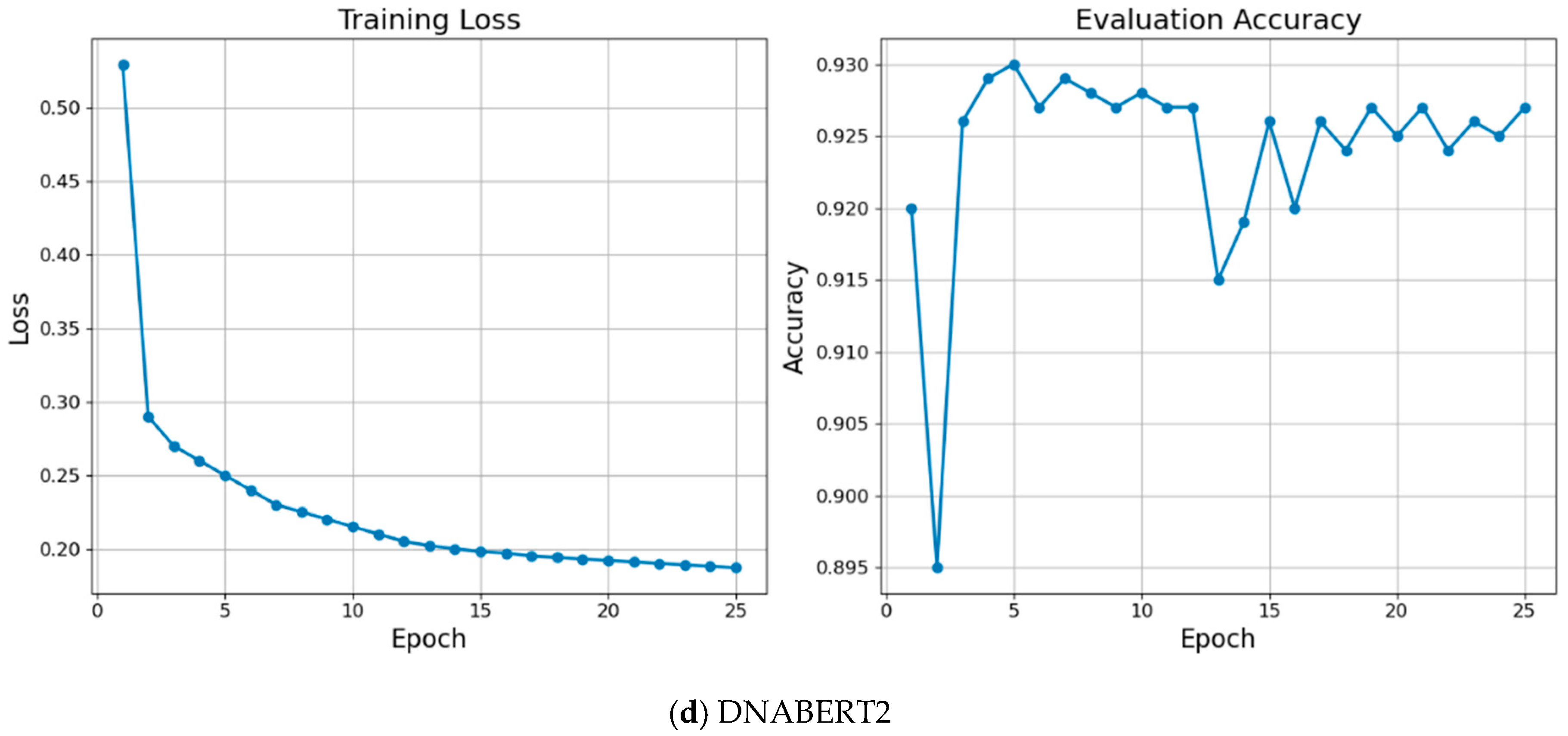
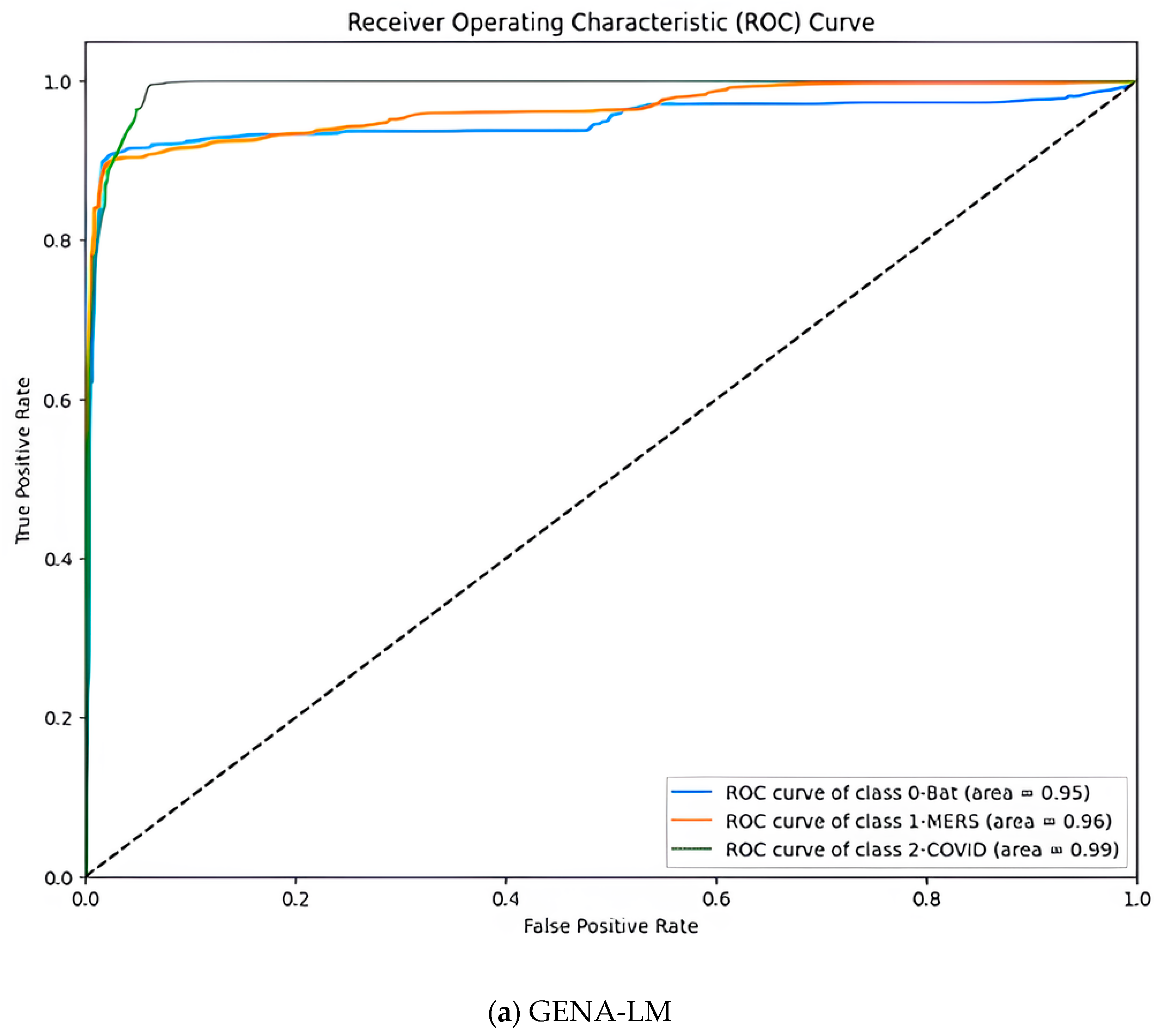
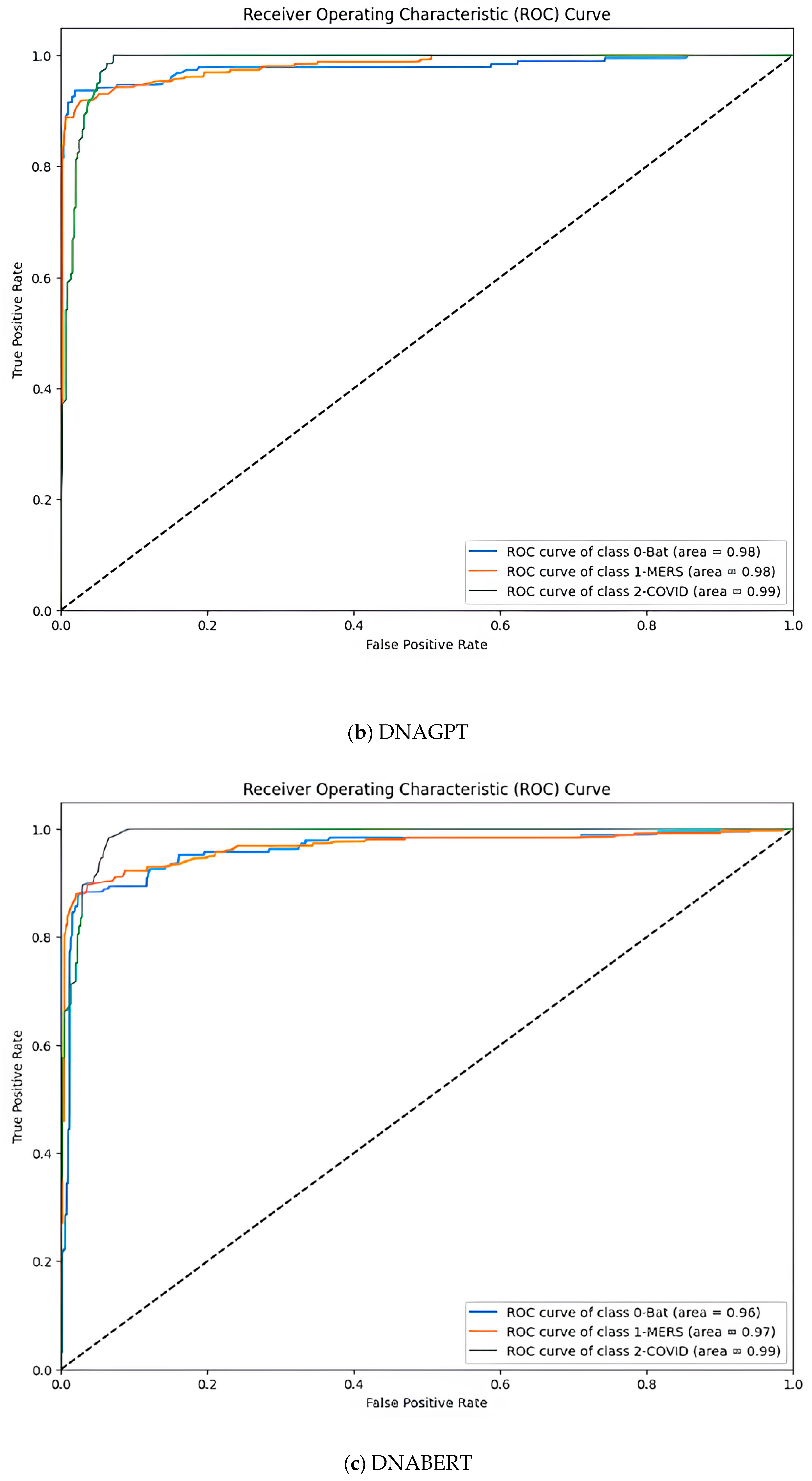
5. Experimental Results and Discussion
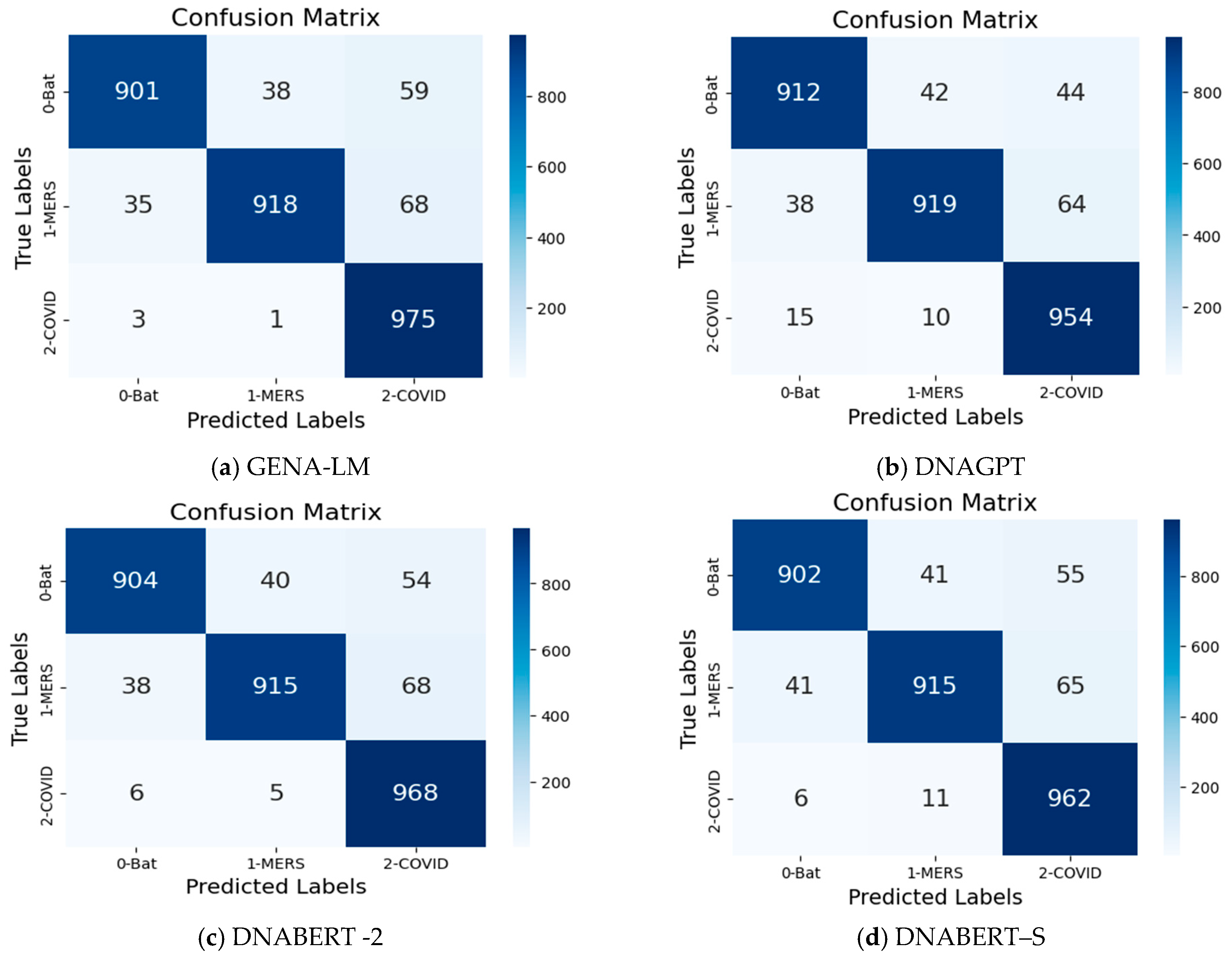
5.1. Model Evaluation and Performance Metrics
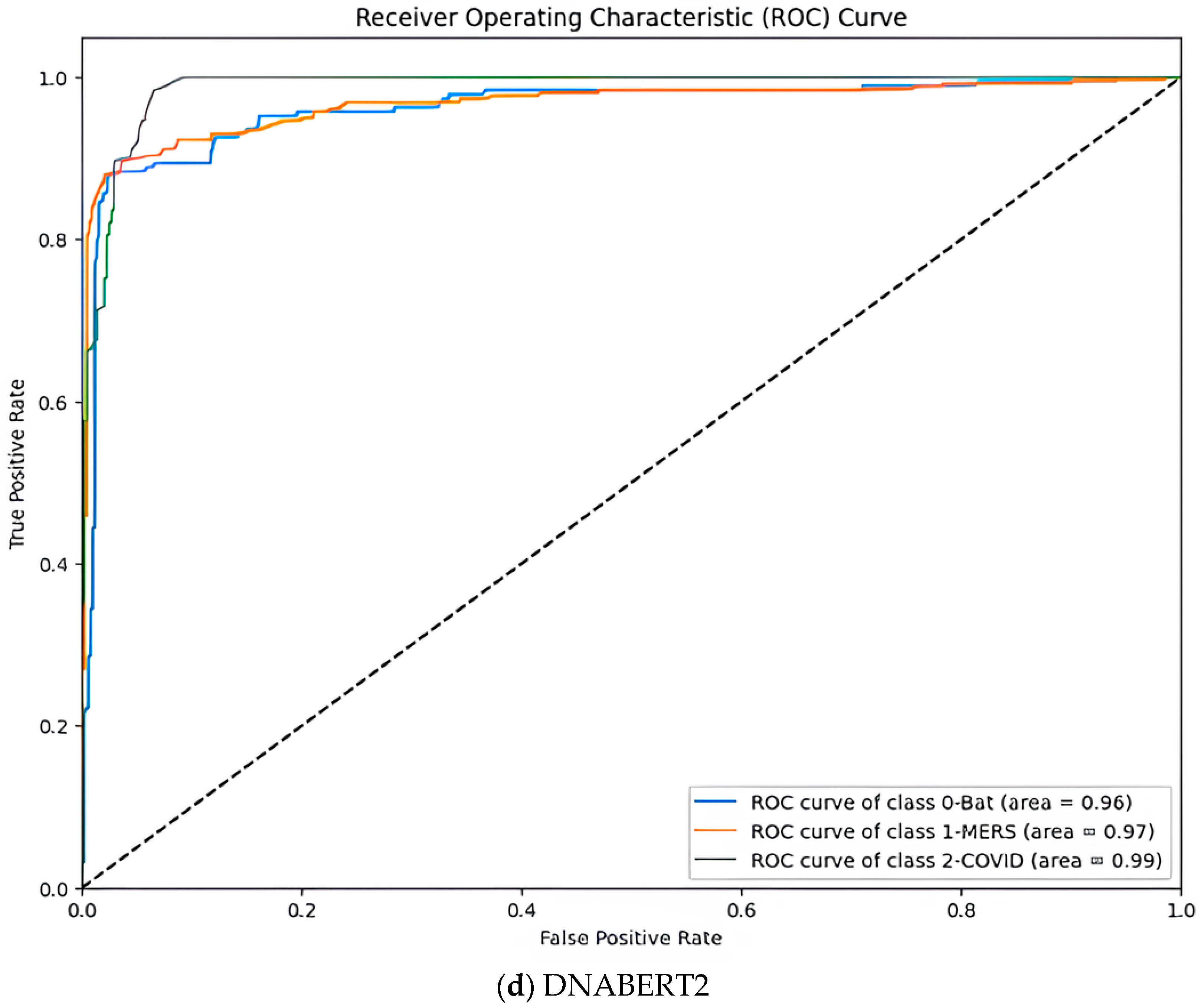
5.2. Model Comparison
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Qayyum, A.; Benzinou, A.; Saidani, O.; Alhayan, F.; Khan, M.A.; Masood, A.; Mazher, M. Assessment and classification of COVID-19 DNA sequence using pairwise features concatenation from multi-transformer and deep features with machine learning models. J. Assoc. Lab. Autom. 2024, 29, 100147. [Google Scholar] [CrossRef]
- Bento, A.I.; Nguyen, T.; Wing, C.; Lozano-Rojas, F.; Ahn, Y.; Simon, K. Evidence from internet search data shows information-seeking responses to news of local COVID-19 cases. Proc. Natl. Acad. Sci. USA 2020, 117, 11220–11222. [Google Scholar] [CrossRef] [PubMed]
- Karlicki, M.; Antonowicz, S.; Karnkowska, A. Tiara: Deep learning-based classification system for eukaryotic sequences. Bioinformatics 2022, 38, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Klapproth, C.; Sen, R.; Stadler, P.F.; Findeiß, S.; Fallmann, J. Common Features in lncRNA Annotation and Classification: A Survey. Non-Coding RNA 2021, 7, 77. [Google Scholar] [CrossRef]
- Yang, A.; Zhang, W.; Wang, J.; Yang, K.; Han, Y.; Zhang, L. Review on the Application of Machine Learning Algorithms in the Sequence Data Mining of DNA. Front. Bioeng. Biotechnol. 2020, 8, 1032. [Google Scholar] [CrossRef]
- Hamed, B.A.; Ibrahim, O.A.S.; El-Hafeez, T.A. Optimizing classification efficiency with machine learning techniques for pattern matching. J. Big Data 2023, 10, 124. [Google Scholar] [CrossRef]
- Rahman, A.; Zaman, S.; Das, D. Cracking the Genetic Codes: Exploring DNA Sequence Classification with Machine Learning Algorithms and Voting Ensemble Strategies. In Proceedings of the 2024 International Conference on Advances in Computing, Communication, Electrical, and Smart Systems (iCACCESS), Dhaka, Bangladesh, 8–9 March 2024; pp. 1–6. [Google Scholar] [CrossRef]
- Nguyen, N.G.; Tran, V.A.; Ngo, D.L.; Phan, D.; Lumbanraja, F.R.; Faisal, M.R.; Abapihi, B.; Kubo, M.; Satou, K. DNA Sequence Classification by Convolutional Neural Network. J. Biomed. Sci. Eng. 2016, 9, 280–286. [Google Scholar] [CrossRef]
- Gomes, J.C.; Masood, A.I.; Silva, L.H.d.S.; Ferreira, J.R.B.d.C.; Júnior, A.A.F.; Rocha, A.L.d.S.; de Oliveira, L.C.P.; da Silva, N.R.C.; Fernandes, B.J.T.; dos Santos, W.P. Covid-19 diagnosis by combining RT-PCR and pseudo-convolutional machines to characterize virus sequences. Sci. Rep. 2021, 11, 11545. [Google Scholar] [CrossRef]
- Gunasekaran, H.; Ramalakshmi, K.; Arokiaraj, A.R.M.; Kanmani, S.D.; Venkatesan, C.; Dhas, C.S.G. Analysis of DNA Sequence Classification Using CNN and Hybrid Models. Comput. Math. Methods Med. 2021, 2021, 1835056. [Google Scholar] [CrossRef]
- Choi, S.R.; Lee, M. Transformer Architecture and Attention Mechanisms in Genome Data Analysis: A Comprehensive Review. Biology 2023, 12, 1033. [Google Scholar] [CrossRef]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. DNABERT: Pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ji, Y.; Li, W.; Dutta, P.; Davuluri, R.; Liu, H. DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genome. arXiv 2023, arXiv:2306.15006v2. [Google Scholar]
- Kabir, A.; Bhattarai, M.; Peterson, S.; Najman-Licht, Y.; Rasmussen, K.Ø.; Shehu, A.; Bishop, A.R.; Alexandrov, B.; Usheva, A. DNA breathing integration with deep learning foundational model advances genome-wide binding prediction of human transcription factors. Nucleic Acids Res. 2024, 52, e91. [Google Scholar] [CrossRef]
- Fishman, V.; Kuratov, Y.; Shmelev, A.; Petrov, M.; Penzar, D.; Shepelin, D.; Chekanov, N.; Kardymon, O.; Burtsev, M. GENA-LM: A family of open-source foundational DNA language models for long sequences. Nucleic Acids Res. 2025, 53, gkae1310. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, W.; Zhao, Y.; Zhang, J.; He, B.; Qin, C.; Yao, J. DNAGPT: A Generalized Pre-trained Tool for Versatile DNA Sequence Analysis Tasks. arXiv 2023, arXiv:2307.05628. [Google Scholar]
- Nguyen, E.; Poli, M.; Faizi, M.; Thomas, A.; Birch-Sykes, C.; Wornow, M.; Patel, A.; Rabideau, C.; Massaroli, S.; Bengio, Y.; et al. HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution. Adv. Neural Inf. Process. Syst. 2023, 36, 43177–43201. [Google Scholar]
- He, S.; Gao, B.; Sabnis, R.; Sun, Q. Nucleic Transformer: Classifying DNA Sequences with Self-Attention and Convolutions. ACS Synth. Biol. 2023, 12, 3205–3214. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Jiang, J.; Chen, P.; Shi, X.; Li, Y. Large Language Models in Bioinformatics: A Survey. arXiv 2025, arXiv:2503.04490. [Google Scholar]
- Sarumi, O.A.; Heider, D. Large language models and their applications in bioinformatics. Comput. Struct. Biotechnol. J. 2024, 23, 3498–3505. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Over-sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Zhang, X.; Beinke, B.; Kindhi, B.A.; Wiering, M. Comparing Machine Learning Algorithms with or without Feature Extraction for DNA Classification. arXiv 2020, arXiv:2011.00485. [Google Scholar]
- Mohammed, R.K.; Alrawi, A.T.H.; Dawood, A.J. U-Net for genomic sequencing: A novel approach to DNA sequence classification. Alex. Eng. J. 2024, 96, 323–331. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| learning rate | 5 × 10−6 |
| training epochs | 10 |
| Optimizer | adamw_torch |
| Stratified K Fold (k-fold) | 10 |
| weight_decay | 0.01 |
| Family | Model | Classes | Performance Metrics | Weighted Average Accuracy | ||
|---|---|---|---|---|---|---|
| Precision | Recall | F1 | ||||
| GENA-LM | BigBird | Bat-Corona | 0.96 | 0.94 | 0.95 | 0.95 |
| MERS | 0.96 | 0.90 | 0.93 | |||
| COVID-2 | 0.91 | 0.99 | 0.95 | |||
| DNAGPT | Bat-Corona | 0.97 | 0.93 | 0.95 | 0.95 | |
| MERS | 0.97 | 0.92 | 0.94 | |||
| COVID-2 | 0.90 | 0.99 | 0.94 | |||
| DNABERT | DNABERT | Bat-Corona | 0.95 | 0.90 | 0.93 | 0.92 |
| MERS | 0.92 | 0.90 | 0.91 | |||
| COVID-2 | 0.89 | 0.96 | 0.92 | |||
| DNABERT-2 | Bat-Corona | 0.95 | 0.93 | 0.94 | 0.94 | |
| MERS | 0.96 | 0.91 | 0.93 | |||
| COVID-2 | 0.91 | 0.98 | 0.95 | |||
| Family | Model | Classes | Performance Metrics | Weighted Average Accuracy | ||
|---|---|---|---|---|---|---|
| Precision | Recall | F1 | ||||
| GENA-LM | BigBird | Bat-Corona | 0.97 | 0.92 | 0.94 | 0.95 |
| MERS | 0.97 | 0.92 | 0.94 | |||
| COVID-2 | 0.90 | 1.00 | 0.95 | |||
| DNAGPT | Bat-Corona | 0.96 | 0.97 | 0.96 | 0.96 | |
| MERS | 0.98 | 0.91 | 0.94 | |||
| COVID-2 | 0.93 | 0.99 | 0.96 | |||
| DNABERT | DNABERT | Bat-Corona | 0.95 | 0.90 | 0.93 | 0.92 |
| MERS | 0.92 | 0.90 | 0.91 | |||
| COVID-2 | 0.89 | 0.96 | 0.92 | |||
| DNABERT-2 | Bat-Corona | 0.96 | 0.94 | 0.95 | 0.95 | |
| MERS | 0.97 | 0.92 | 0.94 | |||
| COVID-2 | 0.92 | 0.99 | 0.95 | |||
| Authors and Years | Method | Dataset | Accuracy |
|---|---|---|---|
| Zhang X et al. [23] | XGBoost | DNA chromosome | 0.88 |
| Hamed, B.A et al. [6] | SVM Sigmoid | DNA dataset | 0.92 |
| Gunasekaran et al. [10] | Deep learning—LSTM | Four-class dataset | 0.93 |
| Shujun He et al. [18] | Nucleic Transformer | E. Coli Classification | 0.88 |
| Zhihan Zhou et al. [13] | DNABERT-2 | Transcription Factor Prediction (Mouse) | 0.92 |
| R. K. Mohammed et al. [24] | U-Net | Three-class dataset | 0.96 |
| Proposed pre-trained LLM | LLM—DNAGPT | Three-class dataset | 0.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunasekaran, H.; Wilfred Blessing, N.R.; Sathic, U.; Husain, M.S. Enhanced Viral Genome Classification Using Large Language Models. Algorithms 2025, 18, 302. https://doi.org/10.3390/a18060302
Gunasekaran H, Wilfred Blessing NR, Sathic U, Husain MS. Enhanced Viral Genome Classification Using Large Language Models. Algorithms. 2025; 18(6):302. https://doi.org/10.3390/a18060302
Chicago/Turabian StyleGunasekaran, Hemalatha, Nesaian Reginal Wilfred Blessing, Umar Sathic, and Mohammad Shahid Husain. 2025. "Enhanced Viral Genome Classification Using Large Language Models" Algorithms 18, no. 6: 302. https://doi.org/10.3390/a18060302
APA StyleGunasekaran, H., Wilfred Blessing, N. R., Sathic, U., & Husain, M. S. (2025). Enhanced Viral Genome Classification Using Large Language Models. Algorithms, 18(6), 302. https://doi.org/10.3390/a18060302