1. Introduction
Cost functions appear in many fields of science, economics, and engineering, wherever issues concerning the optimization, typically the minimization or maximization, of some (scalar) quantity—the so-called cost—are raised [
1]. Classical examples are the cost of a business strategy, the fitness of biological species or individuals, the objective function value of a trajectory in an optimal control problem, or the potential energy of an arrangement of atoms in space. Given a state space of the system of interest, the cost function assigns a real number to each state of the system; such a state is often called a microstate, configuration, or conformation (in the case of chemical systems), or a legitimate solution to a combinatorial optimization problem. Here, the state space can be a discrete set of states, like the nodes of a graph (often called a metagraph in the context of cost function analysis [
2]), or exhibit a continuous structure, like a subspace of
. Together with the neighborhood structure of the state space—in the case of a discrete state space, the connectivity of the metagraph—, the cost function can be represented by a so-called cost function landscape, which usually contains a multitude of local minima that are separated by a complex barrier structure. In this context, one should note that the choice of connectivity or neighborhood is essentially left to the researcher him/herself; since the choice of the neighborhood implicitly defines the dynamics of the system in most cases, one often speaks of the neighborhood in terms of the moveclass of the system—a terminology derived from the description of the exploration algorithms that are designed to study the landscape. Often, the number of local minima grows exponentially or even factorially with some parameter that represents the size of a system, such as the number of cities in a travelling salesman problem [
3,
4], the number of spins in a magnetic material, or the number of atoms in a cluster or molecule [
5]. For more general aspects of cost function, fitness, or energy landscapes, we refer to the literature [
1].
In the case of a chemical or physical system, one frequently encounters the (potential) energy of the system as the quantity to be studied or minimized, and thus one speaks of the energy landscape of, e.g., the molecule of interest. Furthermore, in the case of such systems, the landscape is of much greater importance than just as a tool to find the lowest energy, since the laws of physics that describe the evolution of the system are embedded in the local shape of the potential energy surface (PES): the gradient of the potential energy yields the forces acting on the atoms, and thus prescribes the subsequent trajectory of, e.g., the atoms belonging to a molecule, such as the vibrations or translational motion of the molecule.
Thus, the study of the energy landscape of physical and chemical systems usually goes far beyond the obvious task of identifying the global minimum; for example, all low-energy local minima surrounded by high enough barriers on the landscape correspond to (meta)stable configurations of, e.g., the molecule, which can be observed or synthesized, in principle, or transformed into each other. Therefore, determining all low-energy minima of a chemical system ranks as a goal of similar importance as finding the global minimum [
1].
Furthermore, after the initial step of finding not only the global minimum but also as many side-minima as possible, one would study the energy barriers and entropy barriers of the system separating these minima. This requires the design and implementation of new classes of algorithms that, e.g., identify saddle points between local minima [
6,
7], or measure the entropic barriers between basins for portions of the landscape restricted to lie below energy lids, such as the threshold algorithm [
8].
However, in a large number of instances, just identifying the global minimum without any prior information about the cost function landscape is already a highly nontrivial task that typically requires an enormous computational effort. This is due to the large size of the state space—prohibiting an exhaustive search via testing every microstate—and due to the complexity of the barrier structure, which makes straightforward steepest descent gradient-based minimizations (for continuous landscapes) or random downhill walks ineffective. Although some improvement is achieved by combining such greedy downhill algorithms with a systematic (nearly exhaustive) grid-like selection of starting points for the local minimization [
9], this combination is still not very efficient in identifying the global minimum of the system, due to the enormous size of the state space and the large number of local minima present. Moreover, many of these minima would be rediscovered many times, thus resulting in a massive oversampling of the landscape’s minima.
As a consequence, researchers in many fields have developed a plethora of global optimization algorithms, many of which are variations of some “classical” ones, such as genetic algorithms (GA) [
10], simulated annealing (SA) [
11], or branch-and-bound algorithms (BB) [
12]. Among these are the bounce algorithm [
13], the great-deluge algorithm [
14], threshold accepting [
15], particle swarm optimization [
16], the tabu algorithm [
17], thermal cycling [
18], and evolutionary algorithms [
19,
20,
21], just to name a few—for an overview, we refer to the literature [
1,
22,
23,
24]. Many of these methods are inspired by physical processes, e.g., molecular dynamics simulations [
25,
26] or thermal cycling [
18], and by biology, e.g., the evolutionary [
19] or genetic algorithms [
10,
27], while others are based on general energy landscape considerations as, e.g., the great-deluge [
14] or the tabu [
17] algorithms.
These basic—in principle generally applicable—algorithms are often specially adapted to certain classes of systems and energy landscapes, e.g., for the prediction of the conformers of molecules, such as the rotamers of chain-like molecules such as proteins or polysaccharides. Since one usually restricts the structure generation to molecules with a given bond network, only limited changes in the bond lengths are allowed, and the main degrees of freedom that are varied during the search are the bond angles and dihedral (torsion) angles. Ideally, one would compute the energy on the ab initio level. However, because of the high computational expense of quantum mechanical calculations, one commonly employs empirical molecular modeling potentials instead, possibly adding penalty terms that restrict the bond lengths and angles to certain “allowed” ranges which are physically and chemically sensible—according to experimental data and/or results from ab initio calculations. Such limits can be either implemented as parts of the energy function or via the moveclass, i.e., no new test configuration is allowed that violates these constraints. Moveclasses with such constraints on the allowed configurations are sometimes called “rule-based conformation generators” [
24]; they are employed in the context of molecular structure prediction by, e.g., the OMEGA [
28] method; for a review of these and other approaches, we refer to the literature [
24].
Trying to find the optimal combination of computational speed, quality of the candidates—both regarding their energy and the accuracy of the parameters of the physical structure obtained—, and diversity of the solutions in the set of generated candidates, is a great challenge for systems that exhibit complex multi-minima landscapes. In particular, the goal to explore a wide range of the state space without losing much computational time by having to laboriously cross many energetic barriers as in, e.g., MD simulations, has led to the introduction of algorithms that attempt to perform large moves on the landscape without wasting too much time in exploring uninteresting high-energy regions of the landscape. Such so-called jumpmoves are common features of many modern algorithms, and an optimal choice of the moveclass is crucial for the success of a global optimization procedure [
8]; for example, one class of searches which combines every jumpmove automatically with a local deterministic (gradient-based) or stochastic (downhill random walk) minimization, and therefore moves from local minimum to local minimum, has been called the basin-hopping algorithm (BH) [
29,
30]. Similarly, the genetic algorithms, which belong to the class of multi-walker algorithms that generate new states from two (or more) old states by combining their parameter values, are nowadays often combined with a local minimization of the newly generated state [
31]. Nevertheless, oversampling is still a major problem. Even though the multiple discovery of the same minima is often interpreted as the heuristic criterion of the success (sometimes called “convergence”) of the global optimization, thus turning oversampling from a bug into a feature, performing orders of magnitude more local optimizations than necessary greatly reduces the efficiency of the search algorithm.
Due to the wide variety of fields where global optimizations of cost functions take place, many of these algorithms have been re-invented, or have been transferred from one area of applications to another, where one often has to adapt the original algorithm to the specific aspects of the new class of systems under consideration. An example of such a transfer with adaptation is the application of the RRT (Rapidly exploring Random Tree) algorithm [
32], which was developed in the field of robotics to solve the so-called motion planning problem [
33], to the field of chemistry. Indeed, solving the robot motion planning problem requires algorithms to explore the state space of the robot system aiming to find feasible trajectories between an initial and a final state. The same type of algorithms, with some adaptations, can be applied to explore the energy landscape of atomic or molecular systems [
34].
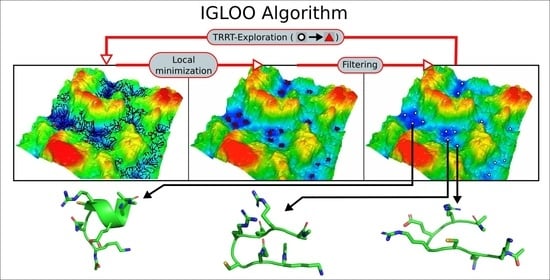
Of particularly great potential to yield a qualitative and not only quantitative improvement in the development of global optimization algorithms are those methods which combine tools and (sub)-algorithms from different fields into a new type of algorithm. In this study, we present the IGLOO (Iterative Global Exploration and Local Optimization) algorithm, which combines concepts from the RRT algorithm and the threshold algorithm to efficiently explore the low-energy regions of the landscape. In the past, an earlier version of this algorithm has been successfully applied to the study of disaccharide molecules on metal surfaces, predicting the shape of the molecules on the surface and thus allowing an interpretation of the STM (Scanning Tunneling Microscopy) images obtained in the experiment [
35].
In the present investigation, we analyze the performance of this algorithm in detail, using two chain-type molecules as examples. The outcomes of these global optimizations are compared to the performance of two similar types of algorithms, a classical basin-hopping algorithm [
36] and a hybrid BH-RRT algorithm [
37].
3. Results and Discussion
To analyse the performance of the algorithms, two molecules with different size and conformational behavior were chosen, as they allow different properties to be evaluated. The first molecule, Met-enkephalin, is an endogenous opioid pentapeptide (Tyr-Gly-Gly-Phe-Met) exhibiting a stable conformational state and various metastable ones [
42]. The second molecule, referred to as df-c-Myb in the following, is an heptapeptide (Ace-Lys-Gln-Cys-Arg-Glu-Arg-Ala-NMe) derived from the recognition helix of the c-Myb DNA-binding protein [
43]. It presents a more complex PES containing regions of the conformational space characterised by different structural motifs [
44]. A good characterization of the PES of these two molecules therefore requires an algorithm able to accurately locate the global minimum while exhaustively exploring the diversity of local minima, both with a high convergence rate. In the following, the performance of the three algorithms presented in
Section 2.1 will be compared on the basis of (
i) their convergence towards the global energy minimum, (
) the associated atomic structures and computing costs, and (
) their ability to explore different regions of the conformational space of the df-c-Myb PES corresponding to three different structural motifs: an
-helix (HLX) and two
-hairpins (HPN1 and HPN2). Note that we have used parallelized versions of the three algorithms. Therefore, the computing times mentioned below correspond to the sum of the CPU time for each thread. Given that we used 45 threads and that the speedup of the parallel version is almost linear with the number of threads, the wall-clock time is approximately the total CPU time divided by 45.
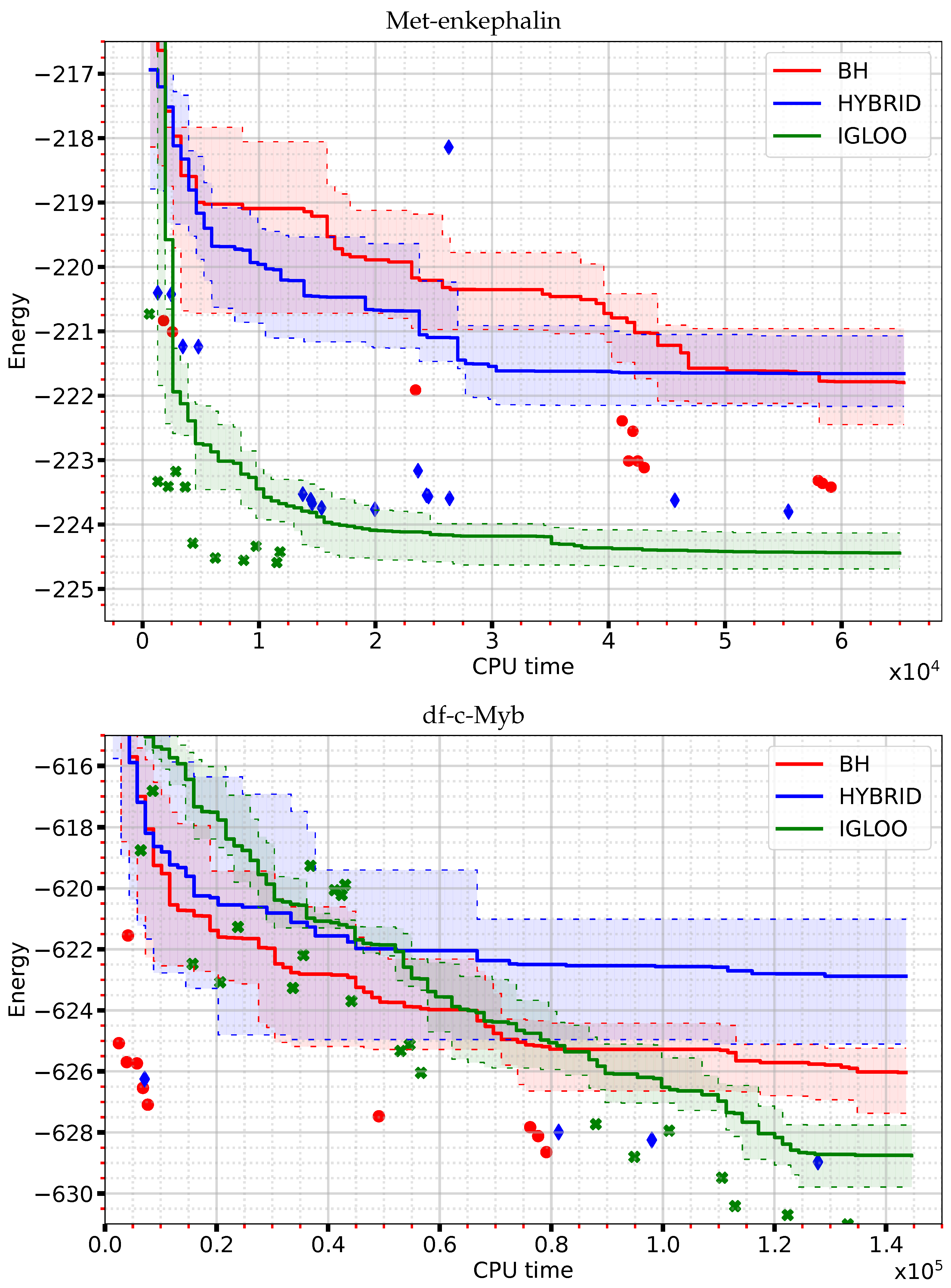
Figure 1 shows the evolution of the observed minimum energy as a function of the CPU time for the molecules used in our analyses. Here we depict, as a function of time, the value of the minimum energy averaged over 10 runs, together with a quartile-based estimate of the spread of the minimum energies observed. In the plot at the top, corresponding to Met-enkephalin, the IGLOO curve remains below the BH and HYBRID ones throughout the exploration. The IGLOO lowest energy decreases very rapidly until around
s, after which the slope decreases markedly and the average energy improves very little beyond
s. The overall behaviour of the HYBRID algorithm is similar to that of IGLOO but with a smaller initial slope and a stagnation of the observed minimum energy after
s. The BH curve looks very different, with a much shallower slope and large plateaus. After
s of exploration, the IGLOO energy (−224.43 kcal/mol) is much lower than the energies of the other two algorithms (−221.63 kcal/mol for HYBRID and −221.77 kcal/mol for BH, respectively). The curves representing the evolution of the lowest energy of df-c-Myb (
Figure 1, bottom panel) show a better performance of IGLOO in terms of convergence towards the minimum energy (−628.75 kcal/mol after
s) compared to BH (−626.04 kcal/mol) and HYBRID (−622.86 kcal/mol). The IGLOO curve has an almost constant slope up to
s, exploration time at which the average minimum energy reaches a plateau. Although its approach to low minimum energies is slower at the beginning, IGLOO surpasses the two other algorithms beyond
s.
The variability of the performance of the algorithms is illustrated by their first and third quartiles shown as dotted lines in
Figure 1. These quartiles were computed from 10 independent runs of the algorithms, using time frames of 650 s for Met-enkephaline and 1450 s for df-c-Myb. For the Met-enkephalin and df-c-Myb, IGLOO exhibits an interquartile mean value of
and
, respectively, vs.
and
, respectively, for HYBRID, and
and
, respectively, for BH. This reflects a lower variability of the results obtained from different executions of IGLOO compared to the two other methods. Due to the stochastic nature of the three algorithms, some runs deviate from their average behavior, as illustrated by the outliers shown in
Figure 1. This confirms the need for a sufficient number of runs to obtain sufficient statistics for a meaningful comparative analysis of these methods. In summary, at short exploration times, the IGLOO algorithm locates structures of lower energy than the BH and HYBRID algorithms and with lower variability, demonstrating its ability to rapidly explore the PES with the objective of finding the global minimum.
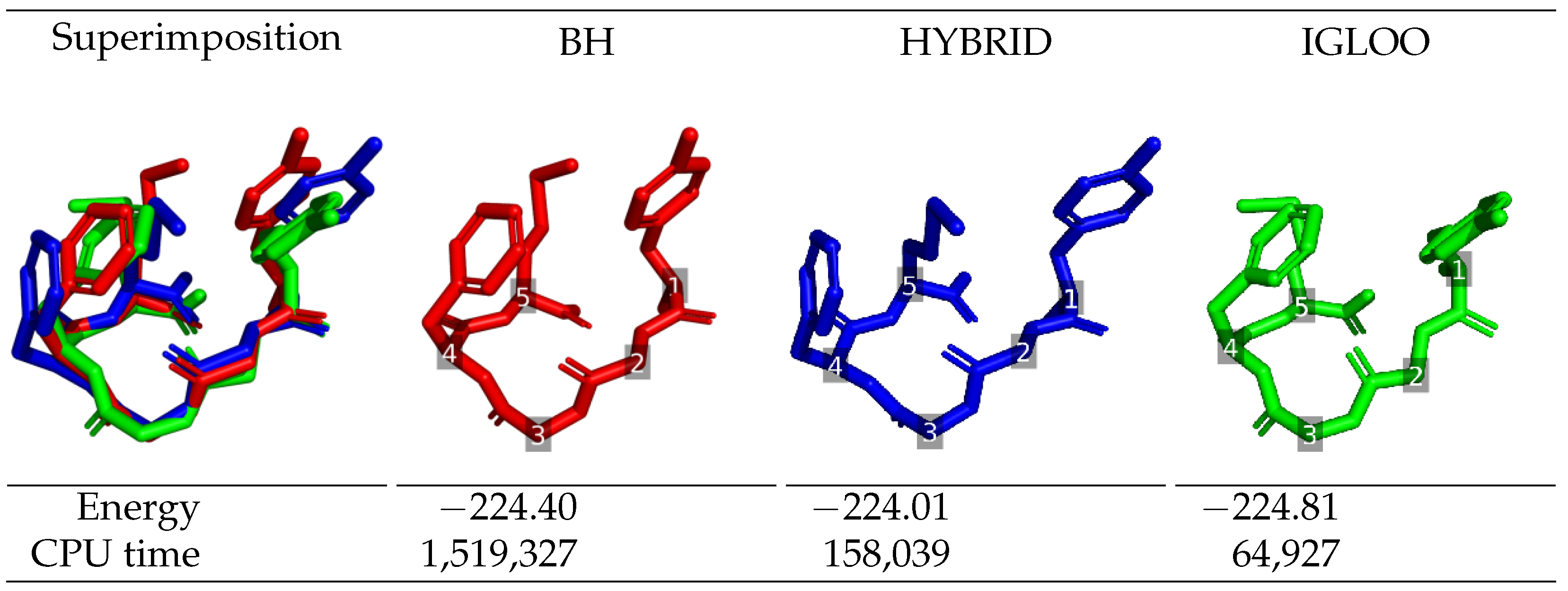
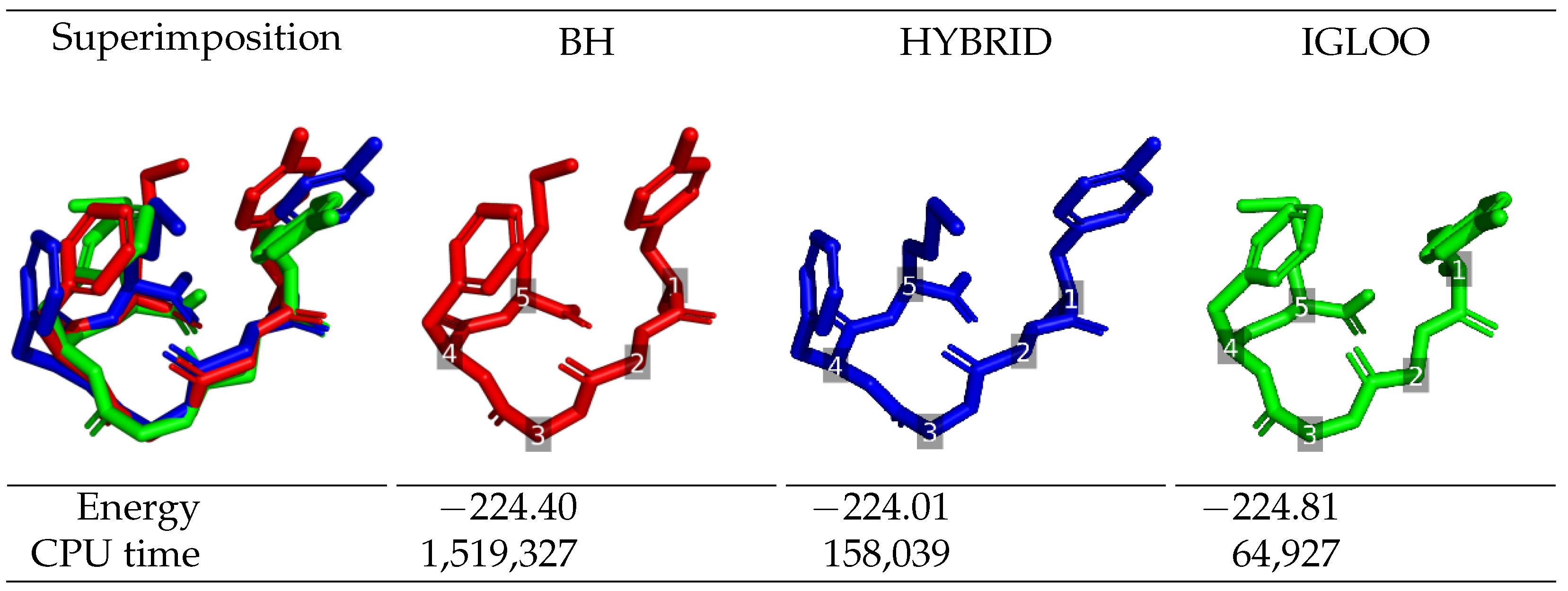
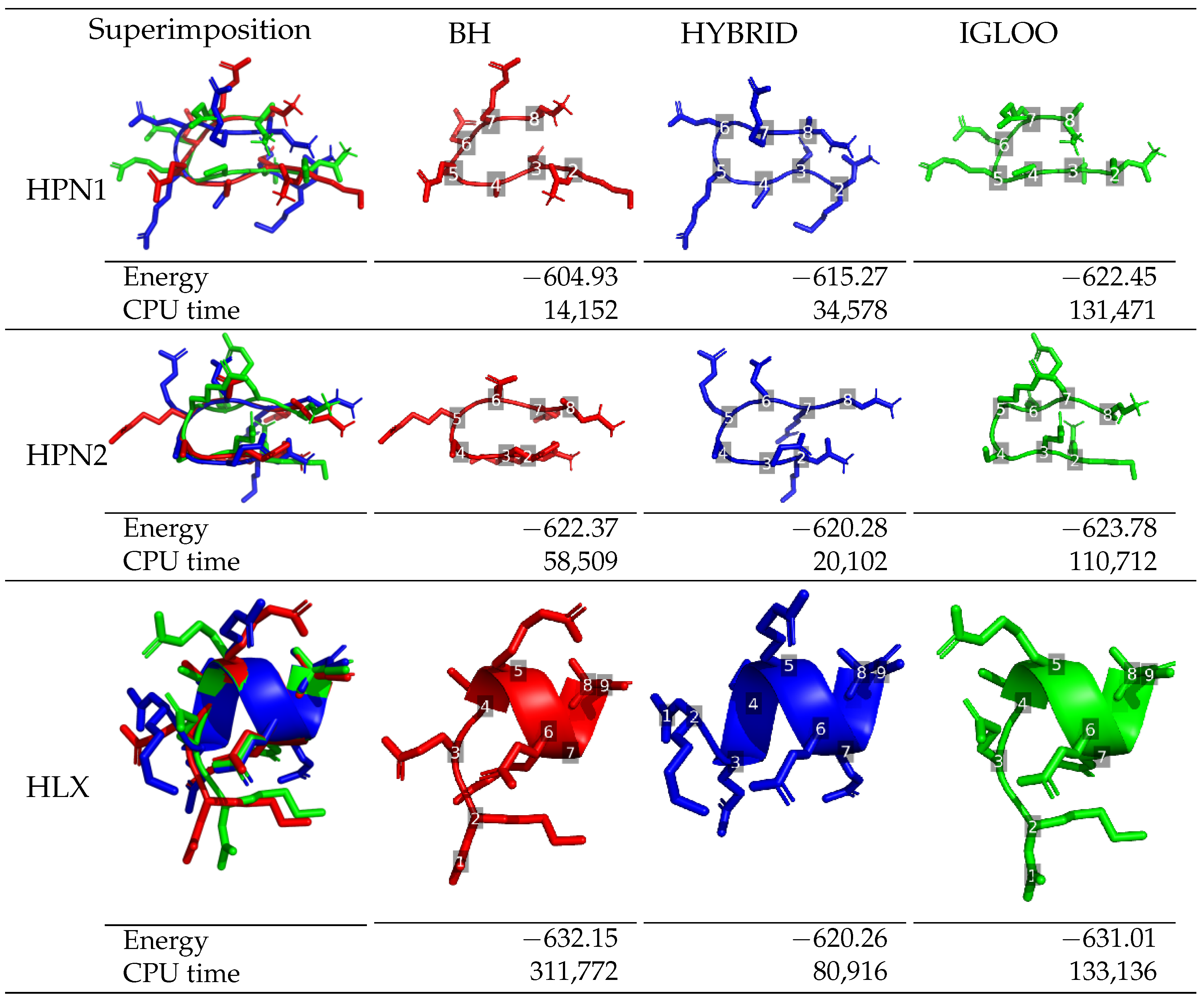
The lowest-energy structures identified by each global optimization method during long runs are depicted in
Figure 2 for Met-enkephalin, and in
Figure 3 for df-c-Myb, together with their energy and the execution time at which they were found. Regarding Met-enkephalin, the structures found by the three algorithms are similar and have very similar energies. The structure obtained by IGLOO was the one with the lowest-energy, followed by BH (+0.41 kcal/mol) and HYBRID (+0.80 kcal/mol). These energy differences come from the side-chain of residue 5 which breaks the stabilizing stacking interaction between the phenol group of residue 1 and the phenyl group of residue 4. The time needed for IGLOO to discover the lowest-energy structure of Met-enkephalin is around 2 and 20 times shorter than the one needed for the HYBRID and BH algorithms, respectively, to locate their optimal structures (which are still slightly less energetically favorable than the one found by IGLOO). This confirms the better performance of IGLOO in terms of convergence to the global minimum.
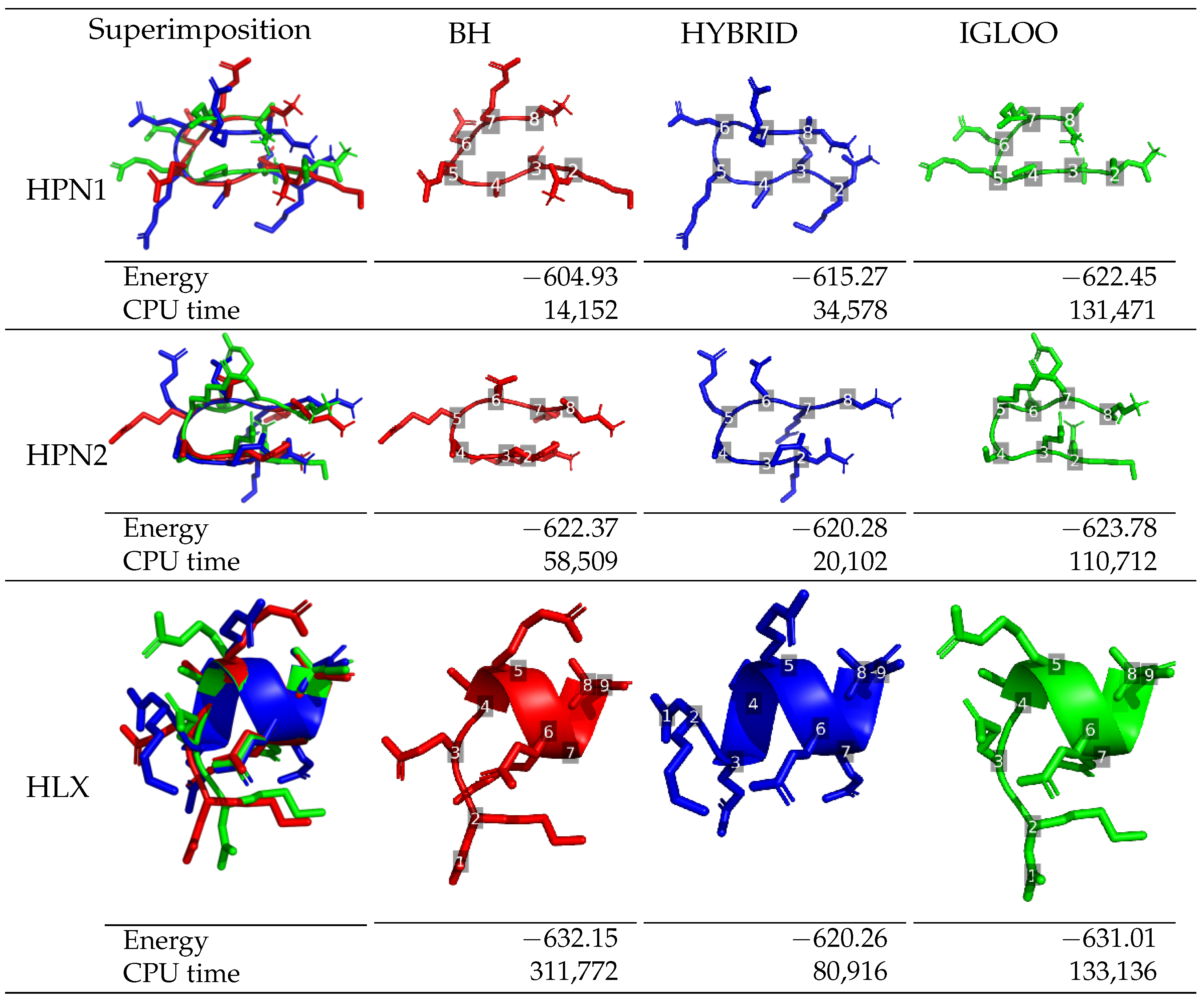
The lowest-energy structures of df-c-Myb corresponding to each of the structural motifs (HPN1, HPN2 and HLX) are presented in
Figure 3 for each of the three algorithms, including the time step when the structures were observed. A secondary structure was classified as a hairpin when it contains a
-turn H-bond (
NH-
O for HPN1 and
NH-
O for HPN2) associated to at least one inter-strand H-bond. The possible inter-strand H-bonds are
NH-
O,
NH-
O,
NH-
O and
NH-
O for HPN1 and
NH-
O,
NH-
O,
NH-
O and
NH-
O for HPN2. A secondary structure was classified as a helix when it contains at least two hydrogen bonds among
NH-
O,
NH-
O,
NH-
O,
NH-
O and
NH-
O. A H-bond was considered to be present when the distance between a donor and an acceptor atom was shorter than a threshold value set to 3.3 Å. The three global optimization methods all succeed in visiting the three structural motif regions: HPN1, HPN2, and HLX. However, the methods differ in their ability to locate the lowest-energy structure for each motif. In the case of hairpins, IGLOO found a more favorable minimum than the ones obtained by BH (+17.52 kcal/mol for HPN1 and +1.41 kcal/mol for HPN2) and the HYBRID algorithm (+7.18 kcal/mol for HPN1 and +3.5 kcal/mol for HPN2). The performance with respect to the low-energy minima exhibiting helices diverges from that of the hairpins, with the BH method finding the best minimum; the best structures found by IGLOO and the HYBRID algorithm having energies 1.14 kcal/mol and 11.89 kcal/mol higher than the best one of the BH structure, respectively. We note that while the BH method finds low-energy helices, it encounters difficulties in the case of hairpins. The HYBRID algorithm explores the different regions of the PES but seems to perform poorly when it comes to refining the explorations locally. Finally, IGLOO manages to quickly identify diverse regions and perform refined local exploration of these regions, where the best minima in each region were found at similar CPU times during the simultaneous multi-basin exploration of the IGLOO runs.
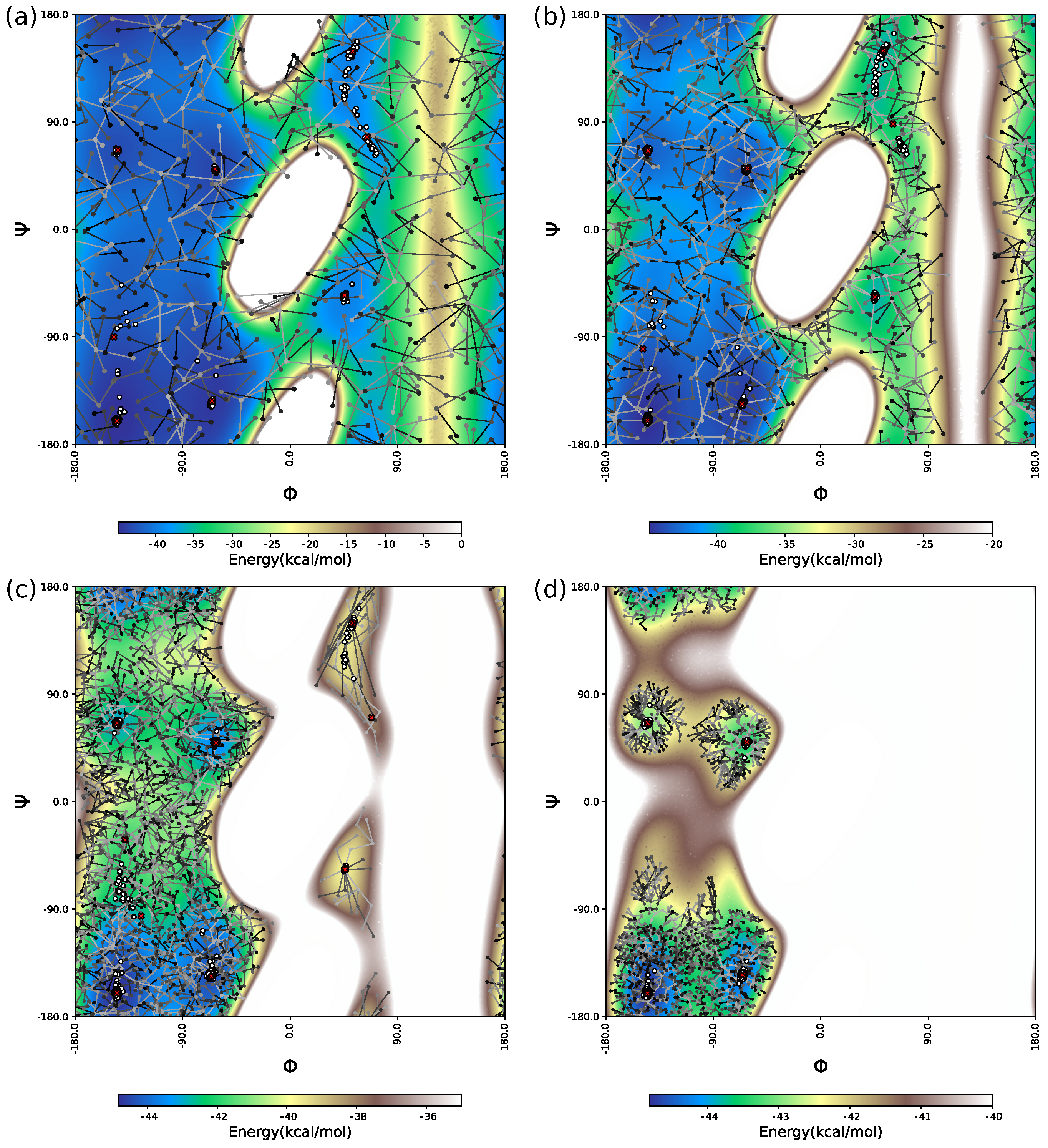
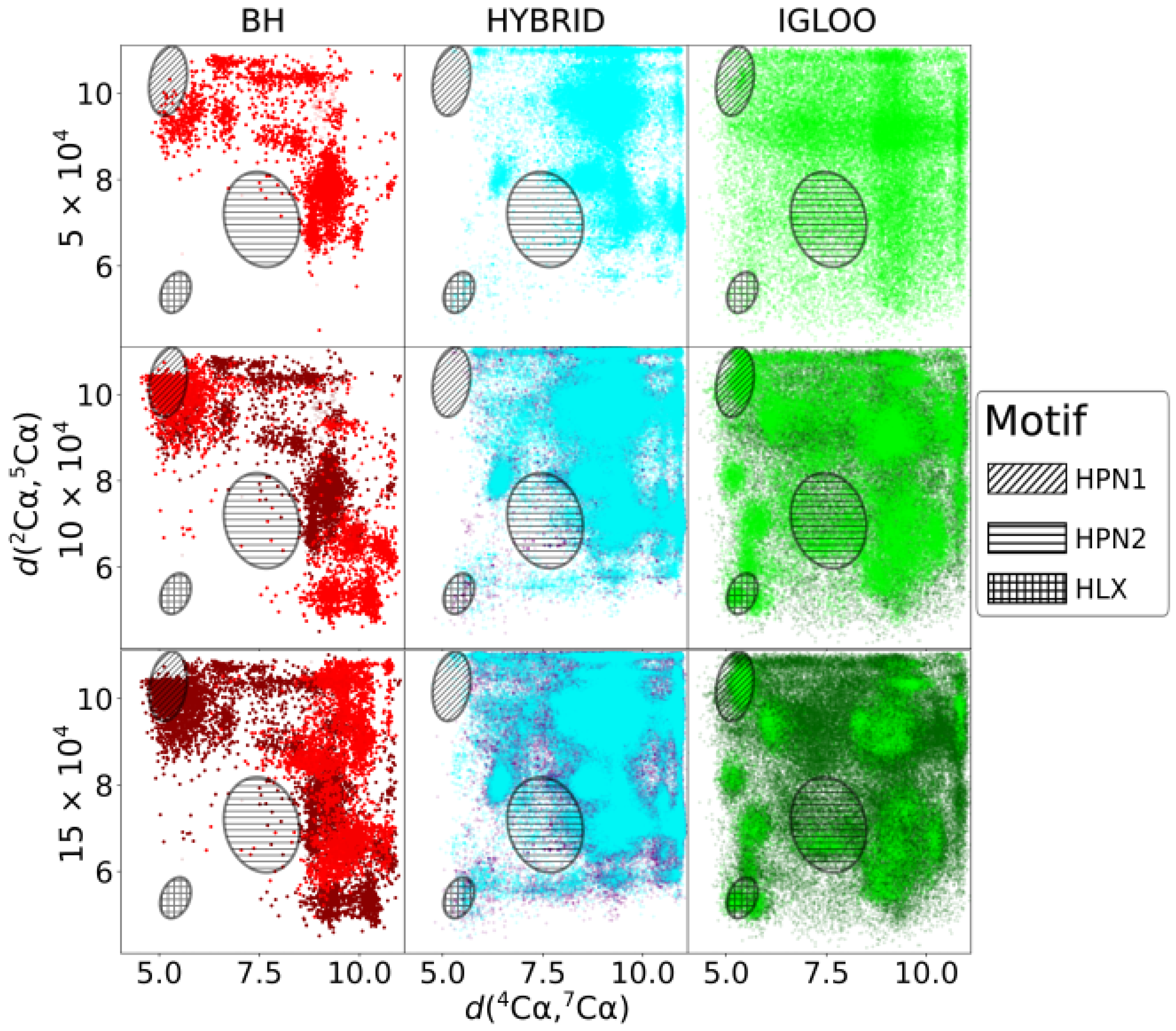
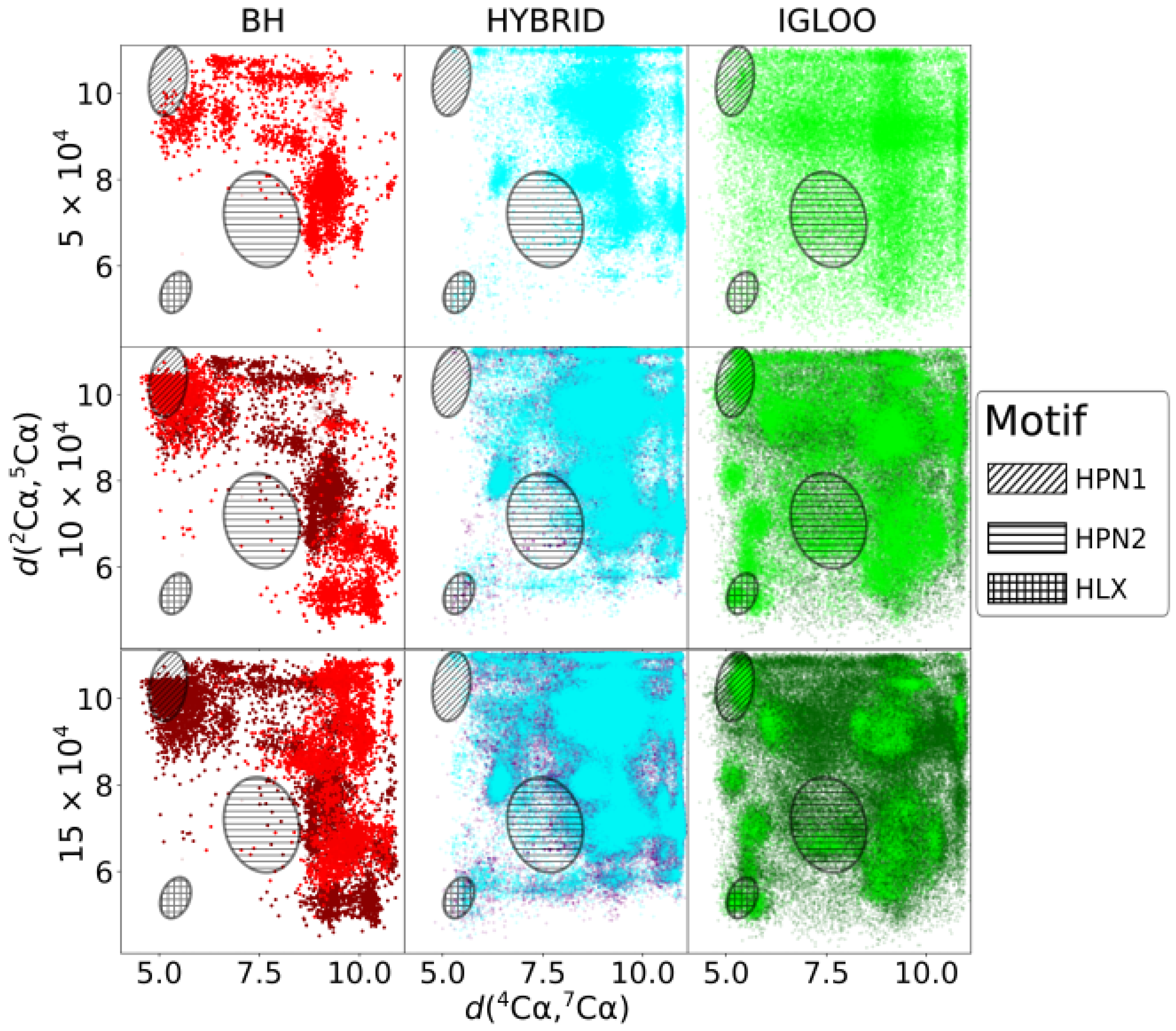
The two-dimensional projection of the minimized conformations issued from df-c-Myb PES explorations is depicted in
Figure 4 for three evenly distributed CPU times (
,
and
s). The two dimensions of the projection, the
C
C
and
C
C
interatomic distances, were chosen because they allow a clear spatial separation of the structural motifs of interest. Regions corresponding to HPN1, HPN2 and HLX structures can thus be approximately delineated on the 2D projection (ovals). The dark dots present on the
and
s snapshots correspond to minima already located at the CPU time of the previous snapshot, i.e.,
and
s respectively.
The BH method struggles to explore the PES of df-c-Myb correctly, with some areas visited very infrequently (HPN2) or not at all (HLX) after
s of exploration, while the others are heavily oversampled. Indeed, very long times are needed to explore the conformational space corresponding to the helices, as shown by the ∼
s needed to locate a low-energy helix (
Figure 3). The BH local exploration also sometimes appears to be of poor quality, as evidenced by the high energy of the most stable HPN1 hairpin found by this algorithm, even though the algorithm has sampled a large part of the corresponding PES region.
The initial space visited by the HYBRID algorithm is more extended than the one visited by the BH method. After
s of exploration, minima are found in the three zones corresponding to the structural motifs. However, during the subsequent exploration, the algorithm concentrates on areas that have already been explored or those that are close to them, and struggles to extend the exploration to previously unvisited regions. Some areas therefore remain unexplored after
s while others appear to be oversampled. A major weakness of the HYBRID algorithm lies in its inability to properly explore locally: although the zones corresponding to the HPN1, HPN2, and HLX motifs are visited, the energies of the corresponding low-energy isomers found by the algorithm remain relatively high (
Figure 3).
Finally, the IGLOO algorithm extends its initial exploration to the whole space and then concentrates on certain zones, including those corresponding to hairpins and helices. Its local refinement is also highly efficient, with the energy of the most stable isomer found by this method being the lowest one in the case of HPN1 and HPN2 and competitive for HLX (
Figure 3). In summary, the BH method and the HYBRID algorithm struggle to cover the space to be explored, oversample certain areas, and show poor performance relative to local refinement. In contrast, the IGLOO algorithm explores the PES in a comprehensive fashion, focusing iteratively on the relevant basins. Furthermore, we note that IGLOO already reaches these relevant basins quite early during the search and thus allows to identify promising low-energy conformations with a rather small computational effort.
4. Conclusions and Outlook
In this study, we have presented a new global optimization algorithm, IGLOO, which combines the basic features of the RRT algorithm from the field of robotics, of the threshold algorithm that has in the past been used to study energy landscapes of various chemical and physical systems, and of repeated local stochastic quenches. Both the RRT and the threshold algorithm explore the landscape in many ways “orthogonal” to the standard global optimization and exploration procedures such as GA, SA, or BH. However, when combined with a moderately greedy local optimization algorithm, we find that IGLOO demonstrates a faster convergence to low-energy minima at a lower computational cost than the other two algorithms we have tested as a comparison (BH and HYBRID), even though they share tools such as frequent minimizations (BH and HYBRID) and the exploration capabilities of the RRT algorithm (HYBRID). In particular, IGLOO achieves a much smoother coverage of all important low-energy regions of the landscape and exhibits a high efficiency in exploring these zones on the landscape. Considering the tools common with the other algorithms, it appears that the threshold criterion likely makes a difference as it tends to focus the search on the low-energy regions of the landscape, without overly constraining the subsequent search inside these regions. In particular, by reducing the importance of lower-level energy barriers but still allowing the RRT-part of the search algorithm to stay in large regions that lie below a (possibly characteristic) threshold energy of the system, the combination of RRT, threshold, and local minimization results in a relatively fast yet efficient and homogeneous coverage of the various low-energy regions of the landscape.
In this “proof-of-principle” study, we have illustrated the ability of IGLOO to identify a representative set of low-energy conformations of flexible peptides. Nevertheless, we can envisage many other applications. A particularly interesting application would be the search for conformers of (small) organic molecules in the context of virtual screening for drug discovery, where IGLOO could be competitive with respect to other algorithms [
24] thanks to its ability to rapidly find diverse low-energy conformations. IGLOO could also be applied to the generation of accessible conformations from a given one with minor modifications of the algorithm, incorporating additional features of the threshold algorithm. Indeed, the main aspect to be changed is the initial iteration of the algorithm, which should start from a given conformation instead of a set of randomly sampled states, and the energy threshold(s) controlling the accessibility should be initialized at the desired value(s). Going further, IGLOO could find applications in many other fields beyond molecular modeling, wherever global optimization methods are useful, including hyperparameter optimization in machine learning [
45]. Note, however, that due to the RRT-based exploration, IGLOO requires the definition of a suitable distance metric in the search space, which is not straightforward for all types of problems.
Concerning potential future applications of IGLOO to various types of chemical systems consisting of one or more molecules, either isolated or on a surface or within a medium, two issues are expected to arise: (i) the quality of the energy function, and (ii) the dimensionality and topology of the configuration space that needs to be explored. Regarding the energy function, full quantum mechanical evaluations of the energy are computationally very expensive and thus always a stretch for global landscape exploration. Nevertheless, energy functions with a good balance between accuracy and computational efficiency, e.g., based on density functional-based tight-binding (DFTB) [
46], are available and have been successfully employed to globally explore energy landscapes of chemical systems [
47].
The issue of the configuration space that needs to be smoothly but efficiently explored is a much more subtle one. We note that it will also appear in any application of IGLOO as a general global optimization algorithm for cost function landscapes drawn not only from robotics and chemistry but also from physics, biology, or economics. The concern here is the question whether the (T)RRT exploration methodology can be employed beyond compact state spaces such as the n-dimensional torus for a chain molecule or a multi-link robot arm where n angular variables characterize the microstates of the system. One class of systems for which (T)RRT appears to be very suitable are the landscapes of periodic approximants to crystalline solids, where the atom coordinates also exhibit a torus-like topology. While the threshold control and the stochastic quenches have proven their worth in many global landscapes studies, testing the (T)RRT feature of IGLOO for other classes of energy landscapes will be an important direction of future research.
The extension of IGLOO to a large variety of cost function problems will, of course, be accompanied by the fine-tuning of the current version of IGLOO, optimizing the parameters of the algorithm and possibly developing adaptive mechanisms for selection and modifying the IGLOO’s control parameters based on information gathered about the landscape. Finally, the graph generation and exploration feature of the (T)RRT part of IGLOO should be a valuable tool for an efficient search for transition path candidates on the energy landscape. Combining this with the measurement of probability flows provided by the threshold algorithm could result in a promising approach for efficiently gaining deeper insights into the barrier structure of an energy landscape, which controls the stability of the minima configurations of a chemical system and governs the transformations among these structures and phases.
Another interesting direction for future work would be the extension of IGLOO to address general problems with high-dimensional state spaces. Up to now, we have successfully tested IGLOO on problems involving several dozen variables, but we can imagine difficulties in tackling problems involving hundreds or thousands of variables, which represent a huge challenge for global exploration. This extension may require a sophisticated parallel implementation of the algorithm. For the investigation presented in this work, we employed a basic multi-threading strategy exploiting the shared-memory architecture of current multi-core CPUs. The execution on larger computer clusters would need a more in-depth strategy, avoiding unnecessary communication between processes. Our previous work on the parallelization of RRT-like algorithms based on an automatic subdivision of space [
48] could be a good starting point. In addition to the “high-level” parallelization of the algorithm for efficient execution on multiple CPUs, one can also envisage GPU-accelerated calculation of certain “low-level” functions inside the algorithm, such as energy or distance calculations. Thus, we expect IGLOO to prove to be a highly versatile tool for global optimization tasks and for the global exploration of complex energy landscapes in the future.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}