
Predicting Dissolution Kinetics of Tricalcium Silicate Using Deep Learning and Analytical Models
 ,
,  ,
,  ,
,
Abstract
1. Introduction

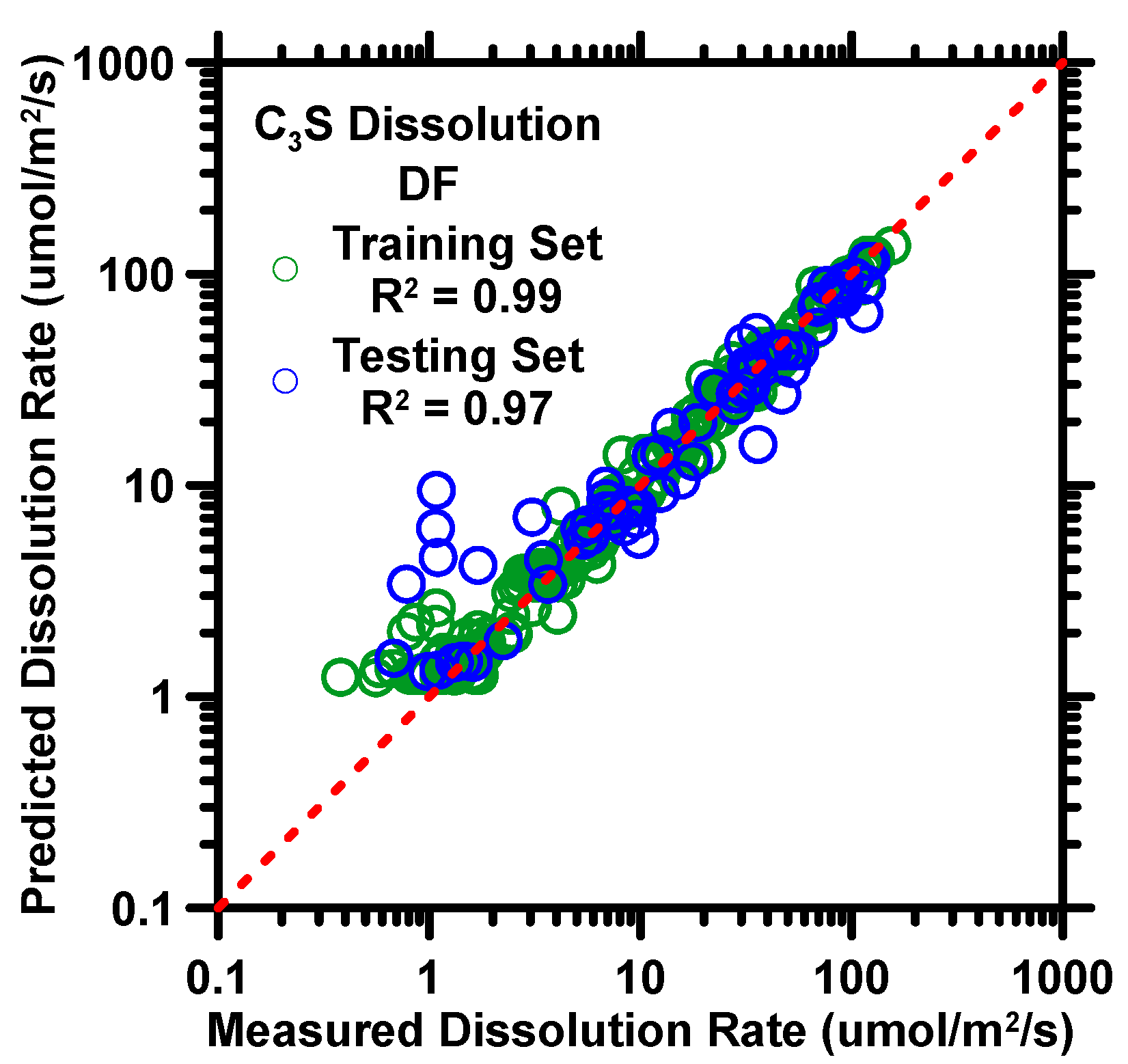{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Burch et al. [27] | |
| Lasaga et al. [28] | |
| Strachan [29] | |
| Oelkers et al. [32] |
2. Database Collection
3. Deep Forest Model
- Each bootstrap iteration in the DF model grows a single tree. At each split, a subset of input variables is randomly selected and used to determine the optimal split scenario. The number of leaves, or the subset size, was set to five in this study. The cost function (i.e., MAE) is used to evaluate all split scenarios, and the scenario with the minimum cost is selected. Unlike other models, the DF model allows trees to grow to their maximum size without pruning or smoothing.
- Next, the DF model produces predictions for OOB data. The DF model aggregates and averages these predictions to produce an overall OOB prediction and OOB error rate. This OOB error rate can be used to evaluate the importance of each variable in influencing the model’s output.
- Lastly, at the testing stage, the DF model averages outcomes from trees to produce predictions for a new data domain.
4. Predictions from Deep Forest Model
5. Analytical Model Development
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gartner, E.; Hirao, H. A Review of Alternative Approaches to the Reduction of CO2 Emissions Associated with the Manufacture of the Binder Phase in Concrete. Cem. Concr. Res. 2015, 78 Pt A, 126–142. [Google Scholar] [CrossRef]
- Schneider, M. Process Technology for Efficient and Sustainable Cement Production. Cem. Concr. Res. 2015, 78 Pt A, 14–23. [Google Scholar] [CrossRef]
- Ludwig, H.-M.; Zhang, W. Research Review of Cement Clinker Chemistry. Cem. Concr. Res. 2015, 78 Pt A, 24–37. [Google Scholar] [CrossRef]
- Bullard, J.W.; Jennings, H.M.; Livingston, R.A.; Nonat, A.; Scherer, G.W.; Schweitzer, J.S.; Scrivener, K.L.; Thomas, J.J. Mechanisms of Cement Hydration. Cem. Concr. Res. 2011, 41, 1208–1223. [Google Scholar] [CrossRef]
- Juilland, P.; Kumar, A.; Gallucci, E.; Flatt, R.J.; Scrivener, K.L. Effect of Mixing on the Early Hydration of Alite and OPC Systems. Cem. Concr. Res. 2012, 42, 1175–1188. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E.; Flatt, R.; Scrivener, K. Dissolution Theory Applied to the Induction Period in Alite Hydration. Cem. Concr. Res. 2010, 40, 831–844. [Google Scholar] [CrossRef]
- Taylor, H.F.W. Cement Chemistry; Thomas Telford: London, UK, 1997. [Google Scholar]
- Oey, T.; Kumar, A.; Falzone, G.; Huang, J.; Kennison, S.; Bauchy, M.; Neithalath, N.; Bullard, J.W.; Sant, G. The Influence of Water Activity on the Hydration Rate of Tricalcium Silicate. J. Am. Ceram. Soc. 2016, 99, 2481–2492. [Google Scholar] [CrossRef]
- Gartner, E.M.; Jennings, H.M. Thermodynamics of Calcium Silicate Hydrates and Their Solutions. J. Am. Ceram. Soc. 1987, 70, 743–749. [Google Scholar] [CrossRef]
- Gartner, E.; Gaidis, J.M. Hydration Mechanisms. In Materials Science of Concrete; Skalny, J.P., Ed.; The American Ceramic Society: Westerville, OH, USA, 1989. [Google Scholar]
- Brown, P.W.; Franz, E.; Frohnsdorff, G.; Taylor, H.F.W. Analyses of the Aqueous Phase during Early C3S Hydration. Cem. Concr. Res. 1984, 14, 257–262. [Google Scholar] [CrossRef]
- Tadros, M.E.; Skalny, J.; Kalyoncu, R.S. Early Hydration of Tricalcium Silicate. J. Am. Ceram. Soc. 1976, 59, 344–347. [Google Scholar] [CrossRef]
- Cabrera, N.; Levine, M.M. XLV. On the Dislocation Theory of Evaporation of Crystals. Philos. Mag. 1956, 1, 450–458. [Google Scholar] [CrossRef]
- Dove, P.M.; Han, N.; De Yoreo, J.J. Mechanisms of Classical Crystal Growth Theory Explain Quartz and Silicate Dissolution Behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 15357–15362. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.L.; McKenna, G.B. The Melting Behavior of Organic Materials Confined in Porous Solids. J. Chem. Phys. 1990, 93, 9002–9011. [Google Scholar] [CrossRef]
- Perez, M. Gibbs–Thomson Effects in Phase Transformations. Scr. Mater. 2005, 52, 709–712. [Google Scholar] [CrossRef]
- Cailleteau, C.; Angeli, F.; Devreux, F.; Gin, S.; Jestin, J.; Jollivet, P.; Spalla, O. Insight into Silicate-Glass Corrosion Mechanisms. Nat. Mater. 2008, 7, 978–983. [Google Scholar] [CrossRef]
- Anbeek, C. Surface Roughness of Minerals and Implications for Dissolution Studies. Geochim. Cosmochim. Acta 1992, 56, 1461–1469. [Google Scholar] [CrossRef]
- Brantley, S.L. Kinetics of Mineral Dissolution. In Kinetics of Water-Rock Interaction; Springer: Berlin, Germany, 2008; pp. 151–210. [Google Scholar] [CrossRef]
- Nicoleau, L.; Nonat, A. A New View on the Kinetics of Tricalcium Silicate Hydration. Cem. Concr. Res. 2016, 86, 1–11. [Google Scholar] [CrossRef]
- Marchon, D.; Juilland, P.; Gallucci, E.; Frunz, L.; Flatt, R.J. Molecular and Submolecular Scale Effects of Comb-Copolymers on Tri-Calcium Silicate Reactivity: Toward Molecular Design. J. Am. Ceram. Soc. 2017, 100, 817–841. [Google Scholar] [CrossRef]
- Fierens, P.; Kabuema, Y.; Tirlocq, J. Influence de La Temperature de Recuit Sur La Cinetique de l’hydratation Du Silicate Tricalcique. Cem. Concr. Res. 1982, 12, 455–462. [Google Scholar] [CrossRef]
- Fischer, C.; Luttge, A. Pulsating Dissolution of Crystalline Matter. Proc. Natl. Acad. Sci. USA 2018, 115, 897–902. [Google Scholar] [CrossRef]
- Casey, W.; Westrich, H. Control of Dissolution Rates of Orthosilicate Minerals by Divalent Metal–Oxygen Bonds. Nature 1992, 355, 157–159. [Google Scholar] [CrossRef]
- Ohlin, C.A.; Villa, E.M.; Rustad, J.R.; Casey, W.H. Dissolution of Insulating Oxide Materials at the Molecular Scale. Nat. Mater. 2010, 9, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lüttge, A. Aluminosilicate Dissolution Kinetics: A General Stochastic Model. J. Phys. Chem. B 2008, 112, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Burch, T.E.; Nagy, K.L.; Lasaga, A.C. Free Energy Dependence of Albite Dissolution Kinetics at 80 °C and PH 8.8. Chem. Geol. 1993, 105, 137–162. [Google Scholar] [CrossRef]
- Lasaga, A.C. Kinetic Theory in the Earth Sciences; Princeton University Press: Princeton, NJ, USA, 1998. [Google Scholar]
- Strachan, D. Glass Dissolution Asa Function of PH and Its Implications for Understanding Mechanisms and Future Experiments. Geochim. Cosmochim. Acta 2017, 219, 111–123. [Google Scholar] [CrossRef]
- Ganor, J.; Lasaga, A.C. Simple Mechanistic Models for Inhibition of a Dissolution Reaction. Geochim. Cosmochim. Acta 1998, 62, 1295–1306. [Google Scholar] [CrossRef]
- Lasaga, A.C. Chapter 2. Fundamental Approaches in Describing Mineral Dissolution and Precipitation Rates. In Chemical Weathering Rates of Silicate Minerals; White, A.F., Brantley, S.L., Eds.; De Gruyter: Berlin, Germany, 1995; pp. 23–86. [Google Scholar]
- Oelkers, E.H.; Schott, J.; Devidal, J.-L. The Effect of Aluminum, PH, and Chemical Affinity on the Rates of Aluminosilicate Dissolution Reactions. Geochim. Cosmochim. Acta 1994, 58, 2011–2024. [Google Scholar] [CrossRef]
- Oelkers, E.H. General Kinetic Description of Multioxide Silicate Mineral and Glass Dissolution. Geochim. Cosmochim. Acta 2001, 65, 3703–3719. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Schott, J. An Experimental Study of Enstatite Dissolution Rates as a Function of PH, Temperature, and Aqueous Mg and Si Concentration, and the Mechanism of Pyroxene/Pyroxenoid Dissolution. Geochim. Cosmochim. Acta 2001, 65, 1219–1231. [Google Scholar] [CrossRef]
- Hellmann, R. The Albite-Water System: Part II. The Time-Evolution of the Stoichiometry of Dissolution as a Function of pH at 100, 200, and 300 °C. Geochim. Cosmochim. Acta 1995, 59, 1669–1697. [Google Scholar] [CrossRef]
- Brantley, S.L.; Stillings, L. Feldspar Dissolution at 25 °C and Low pH. Am. J. Sci. 1996, 296, 101–127. [Google Scholar] [CrossRef]
- Nicoleau, L.; Nonat, A.; Perrey, D. The Di- and Tricalcium Silicate Dissolutions. Cem. Concr. Res. 2013, 47, 14–30. [Google Scholar] [CrossRef]
- Nicoleau, L.; Schreiner, E.; Nonat, A. Ion-Specific Effects Influencing the Dissolution of Tricalcium Silicate. Cem. Concr. Res. 2014, 59, 118–138. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E. Morpho-Topological Investigation of the Mechanisms and Kinetic Regimes of Alite Dissolution. Cem. Concr. Res. 2015, 76, 180–191. [Google Scholar] [CrossRef]
- Han, T.; Stone-Weiss, N.; Huang, J.; Goel, A.; Kumar, A. Machine Learning as a Tool to Design Glasses with Controlled Dissolution for Application in Healthcare Industry. Acta Biomater. 2020, 107, 286–298. [Google Scholar] [CrossRef]
- Cook, R.; Lapeyre, J.; Ma, H.; Kumar, A. Prediction of Compressive Strength of Concrete: A Critical Comparison of Performance of a Hybrid Machine Learning Model with Standalone Models. ASCE J. Mater. Civ. Eng. 2019, 31, 04019255. [Google Scholar] [CrossRef]
- Han, T.; Siddique, A.; Khayat, K.; Huang, J.; Kumar, A. An Ensemble Machine Learning Approach for Prediction and Optimization of Modulus of Elasticity of Recycled Aggregate Concrete. Constr. Build. Mater. 2020, 244, 118271. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, T.; Anoop Krishnan, N.M.; Smedskjaer, M.M.; Ryan, J.V.; Gin, S.; Bauchy, M. Predicting the Dissolution Kinetics of Silicate Glasses by Topology-Informed Machine Learning. npj Mater. Degrad. 2019, 3, 32. [Google Scholar] [CrossRef]
- Chou, J.-S.; Tsai, C.-F. Concrete Compressive Strength Analysis Using a Combined Classification and Regression Technique. Autom. Constr. 2012, 24, 52–60. [Google Scholar] [CrossRef]
- Omran, B.A.; Chen, Q.; Jin, R. Comparison of Data Mining Techniques for Predicting Compressive Strength of Environmentally Friendly Concrete. J. Comput. Civ. Eng. 2016, 30, 04016029. [Google Scholar] [CrossRef]
- Duan, Z.H.; Kou, S.C.; Poon, C.S. Using Artificial Neural Networks for Predicting the Elastic Modulus of Recycled Aggregate Concrete. Constr. Build. Mater. 2013, 44, 524–532. [Google Scholar] [CrossRef]
- Bangaru, S.S.; Wang, C.; Hassan, M.; Jeon, H.W.; Ayiluri, T. Estimation of the Degree of Hydration of Concrete through Automated Machine Learning Based Microstructure Analysis—A Study on Effect of Image Magnification. Adv. Eng. Inform. 2019, 42, 100975. [Google Scholar] [CrossRef]
- Gomaa, E.; Han, T.; ElGawady, M.; Huang, J.; Kumar, A. Machine Learning to Predict Properties of Fresh and Hardened Alkali-Activated Concrete. Cem. Concr. Compos. 2021, 115, 103863. [Google Scholar] [CrossRef]
- Elçiçek, H.; Akdoğan, E.; Karagöz, S. The Use of Artificial Neural Network for Prediction of Dissolution Kinetics. Sci. World J. 2014, 2014, e194874. [Google Scholar] [CrossRef]
- Xu, X.; Han, T.; Huang, J.; Kruger, A.A.; Kumar, A.; Goel, A. Machine Learning Enabled Models to Predict Sulfur Solubility in Nuclear Waste Glasses. ACS Appl. Mater. Interfaces 2021, 13, 53375–53387. [Google Scholar] [CrossRef]
- Cook, R.; Han, T.; Childers, A.; Ryckman, C.; Khayat, K.; Ma, H.; Huang, J.; Kumar, A. Machine Learning for High-Fidelity Prediction of Cement Hydration Kinetics in Blended Systems. Mater. Des. 2021, 208, 109920. [Google Scholar] [CrossRef]
- Lapeyre, J.; Han, T.; Wiles, B.; Ma, H.; Huang, J.; Sant, G.; Kumar, A. Machine Learning Enables Prompt Prediction of Hydration Kinetics of Multicomponent Cementitious Systems. Sci. Rep. 2021, 11, 3922. [Google Scholar] [CrossRef]
- Han, T.; Ponduru, S.A.; Cook, R.; Huang, J.; Sant, G.; Kumar, A. A Deep Learning Approach to Design and Discover Sustainable Cementitious Binders: Strategies to Learn from Small Databases and Develop Closed-Form Analytical Models. Front. Mater. 2022, 8, 796476. [Google Scholar] [CrossRef]
- Bellmann, F.; Sowoidnich, T.; Ludwig, H.-M.; Damidot, D. Dissolution Rates During the Early Hydration of Tricalcium Silicate. Cem. Concr. Res. 2015, 72, 108–116. [Google Scholar] [CrossRef]
- Damidot, D.; Bellmann, F.; Sovoidnich, T.; Möser, B. Measurement and Simulation of the Dissolution Rate at Room Temperature in Conditions Close to a Cement Paste: From Gypsum to Tricalcium Silicate. J. Sustain. Cem.-Based Mater. 2012, 1, 94–110. [Google Scholar] [CrossRef]
- Barret, P.; Ménétrier, D. Filter Dissolution of C3S as a Function of the Lime Concentration in a Limited Amount of Lime Water. Cem. Concr. Res. 1980, 10, 521–534. [Google Scholar] [CrossRef]
- Robin, V.; Wild, B.; Daval, D.; Pollet-Villard, M.; Nonat, A.; Nicoleau, L. Experimental Study and Numerical Simulation of the Dissolution Anisotropy of Tricalcium Silicate. Chem. Geol. 2018, 497, 64–73. [Google Scholar] [CrossRef]
- Breiman, L. Bagging Predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, X.; Ishwaran, H. Random Forests for Genomic Data Analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef]
- Ibrahim, I.A.; Khatib, T. A Novel Hybrid Model for Hourly Global Solar Radiation Prediction Using Random Forests Technique and Firefly Algorithm. Energy Convers. Manag. 2017, 138, 413–425. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Carlini, N.; Erlingsson, Ú.; Papernot, N. Distribution Density, Tails, and Outliers in Machine Learning: Metrics and Applications. arXiv 2019. [Google Scholar] [CrossRef]
- Chakravarty, S.; Demirhan, H.; Baser, F. Fuzzy Regression Functions with a Noise Cluster and the Impact of Outliers on Mainstream Machine Learning Methods in the Regression Setting. Appl. Soft Comput. 2020, 96, 106535. [Google Scholar] [CrossRef]
- Schaffer, C. Selecting a Classification Method by Cross-Validation. Mach. Learn. 1993, 13, 135–143. [Google Scholar] [CrossRef]
- Crundwell, F.K. On the Mechanism of the Dissolution of Quartz and Silica in Aqueous Solutions. ACS Omega 2017, 2, 1116. [Google Scholar] [CrossRef] [PubMed]
- Bergstra, J.; Bengio, Y. Random Search for Hyper-Parameter Optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- Dove, P.M.; Han, N. Kinetics of Mineral Dissolution and Growth as Reciprocal Microscopic Surface Processes across Chemical Driving Force. AIP Conf. Proc. 2007, 916, 215. [Google Scholar] [CrossRef]
- Flatt, R.J.; Scherer, G.W.; Bullard, J.W. Why Alite Stops Hydrating below 80% Relative Humidity. Cem. Concr. Res. 2011, 41, 987–992. [Google Scholar] [CrossRef]
- Kumar, A.; Bishnoi, S.; Scrivener, K.L. Modelling Early Age Hydration Kinetics of Alite. Cem. Concr. Res. 2012, 42, 903–918. [Google Scholar] [CrossRef]
- Zhang, Z.; Han, F.; Yan, P. Modelling the Dissolution and Precipitation Process of the Early Hydration of C3S. Cem. Concr. Res. 2020, 136, 106174. [Google Scholar] [CrossRef]
- Bullard, J.W.; Scherer, G.W.; Thomas, J.J. Time Dependent Driving Forces and the Kinetics of Tricalcium Silicate Hydration. Cem. Concr. Res. 2015, 74, 26–34. [Google Scholar] [CrossRef]
- USGS—Description of Input and Examples for PHREEQC Version 3—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. Available online: https://pubs.usgs.gov/tm/06/a43/pdf/tm6-A43.pdf (accessed on 13 November 2022).
- Bothe, J.V.; Brown, P.W. PhreeqC Modeling of Friedel’s Salt Equilibria at 23 ± 1 °C. Cem. Concr. Res. 2004, 34, 1057–1063. [Google Scholar] [CrossRef]
- Halim, C.E.; Short, S.A.; Scott, J.A.; Amal, R.; Low, G. Modelling the Leaching of Pb, Cd, As, and Cr from Cementitious Waste Using PHREEQC. J. Hazard. Mater. 2005, 125, 45–61. [Google Scholar] [CrossRef]
- Benavente, D.; Brimblecombe, P.; Grossi, C.M. Thermodynamic Calculations for the Salt Crystallisation Damage in Porous Built Heritage Using PHREEQC. Environ. Earth Sci. 2015, 74, 2297–2313. [Google Scholar] [CrossRef]
- Friedman, J.H. Stochastic Gradient Boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Lapeyre, J.; Kumar, A. Influence of Pozzolanic Additives on Hydration Mechanisms of Tricalcium Silicate. J. Am. Ceram. Soc. 2018, 101, 3557–3574. [Google Scholar] [CrossRef]
- Meng, W.; Lunkad, P.; Kumar, A.; Khayat, K. Influence of Silica Fume and Polycarboxylate Ether Dispersant on Hydration Mechanisms of Cement. J. Phys. Chem. C 2016, 120, 26814–26823. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- McKinnon, K.I.M. Convergence of the Nelder--Mead Simplex Method to a Nonstationary Point. SIAM J. Optim. 1998, 9, 148–158. [Google Scholar] [CrossRef]





| Attribute | Unit | Min. | Max. | Mean | Std. Dev. |
|---|---|---|---|---|---|
| Temperature | °C | 10 | 60 | 21.07 | 5.437 |
| SSA of C3S | m2/g | 0 | 0.400 | 0.112 | 0.171 |
| Flow Rate | mL/min/mm2 | 0 | 1273 | 79.22 | 201.8 |
| Initial Na Concentration | mM | 0 | 1000 | 29.19 | 101.5 |
| Initial Cl Concentration | mM | 0 | 1000 | 18.74 | 113.6 |
| Initial Ca Concentration | mM | 0 | 20 | 5.824 | 6.561 |
| Initial Si Concentration | mM | 0 | 0.876 | 0.006 | 0.062 |
| Initial Cs Concentration | mM | 0 | 1000 | 5.513 | 65.45 |
| Initial K Concentration | mM | 0 | 1000 | 5.513 | 65.45 |
| Initial SO4 Concentration | mM | 0 | 200 | 8.904 | 34.95 |
| Initial pH | Unitless | 6.516 | 13.09 | 10.69 | 2.316 |
| C3S Dissolution rate | µmol/m2/s | 0.3800 | 154.6 | 27.92 | 32.61 |
| Model Name | R | R2 | MAE | MAPE | RMSE |
| DF | Unitless | Unitless | µmol/m2/s | % | µmol/m2/s |
| 0.9672 | 0.9354 | 5.297 | 47.33 | 9.373 |
| C0 | 59.7404 | C1 | −17.0531 | C2 | −0.3166 |
| C3 | −231.8133 | C4 | 1.7087 | C5 | 1.7798 |
| C6 | 0.0256 | C7 | −0.0646 |
| Model Name | R | R2 | MAE | MAPE | RMSE | |
|---|---|---|---|---|---|---|
| Unitless | Unitless | µmol/m2/s | % | µmol/m2/s | ||
| Generic Solvent | Analytical model | 0.8277 | 0.6851 | 13.76 | 55.05 | 32.90 |
| Alkaline Solvent | Analytical model | 0.9566 | 0.9151 | 4.921 | 39.77 | 9.545 |
| C0 | −1160.8543 | C1 | −1476.3562 | C2 | −0.6632 |
| C3 | −256.4132 | C4 | −37.9113 | C5 | −37.9089 |
| C6 | −0.3445 | C7 | −0.0978 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, T.; Ponduru, S.A.; Reka, A.; Huang, J.; Sant, G.; Kumar, A. Predicting Dissolution Kinetics of Tricalcium Silicate Using Deep Learning and Analytical Models. Algorithms 2023, 16, 7. https://doi.org/10.3390/a16010007
Han T, Ponduru SA, Reka A, Huang J, Sant G, Kumar A. Predicting Dissolution Kinetics of Tricalcium Silicate Using Deep Learning and Analytical Models. Algorithms. 2023; 16(1):7. https://doi.org/10.3390/a16010007
Chicago/Turabian StyleHan, Taihao, Sai Akshay Ponduru, Arianit Reka, Jie Huang, Gaurav Sant, and Aditya Kumar. 2023. "Predicting Dissolution Kinetics of Tricalcium Silicate Using Deep Learning and Analytical Models" Algorithms 16, no. 1: 7. https://doi.org/10.3390/a16010007
APA StyleHan, T., Ponduru, S. A., Reka, A., Huang, J., Sant, G., & Kumar, A. (2023). Predicting Dissolution Kinetics of Tricalcium Silicate Using Deep Learning and Analytical Models. Algorithms, 16(1), 7. https://doi.org/10.3390/a16010007