Abstract
A code MolecGeom, based on algorithms for stepwise distortions of bond lengths, bond angles and dihedral angles of polyatomic molecules, is presented. Potential energy surfaces (PESs) are curated in terms of the energy for each molecular geometry. PESs based on the Born–Oppenheimer approximation, by which the atomic nuclei within a molecule are assumed stationary with respect to the motion of its electrons, are calculated. Applications requiring PESs involve the effects of nuclear motion on molecular properties. These include determining equilibrium geometries corresponding to stationary and saddle point energies, calculating reaction rates and predicting vibrational spectra. This multi-objective study focuses on the development of a new method for the calculation of PESs and the analysis of the molecular geometry components in terms of incremental changes that provide comprehensive sampling while preserving the precision of PES points. MolecGeom is applied to generate geometric data to calculate PESs for theoretical calculations of vibrational-rotational spectra of water and formaldehyde. An ab initio PES comprising 525 and 2160 intramolecular nuclear configurations results in vibrational frequencies in agreement with experiment, having errors less than 0.08% and 0.8%, respectively. Vinyl alcohol, with a total of 14 internal coordinates, generates a PES of 1458 unique geometries. Ascorbic acid, with 54 internal coordinates, generates a 1,899,776 point PES.
1. Introduction
A numerical molecular potential energy surface (PES) can be depicted by calculating total electronic energies as a function of geometric parameters. These include molecular bond lengths, bond angles and dihedral angles defined in an internal coordinate or Z-matrix space. For a nonlinear polyatomic molecule comprised of N atoms, the PES is determined by its 3N-6 internal coordinates [1,2]. For large polyatomic systems there is no single established effective procedure by which geometries for PESs may be generated. The code presented here, based on symbolic computation, enables the generation of comprehensive PESs based on chosen incremental changes in internal coordinates, relative to a referenced geometrical point. Applications of PES’s include the analysis of vibrational-rotational spectra, synthetic mechanistic pathways and chemical kinetics. This study elaborates on the application of PESs to vibrational-rotational spectra and presents such an analysis of the water and formaldehyde molecules.
The ultimate success of this multi-objective study relies on the automation and efficiency afforded by Mathematica while maintaining sound theoretical underpinnings [3]. There are two principal formal and algorithmic components that must be addressed in the analysis of PESs. The first is that the manual solution of the requisite multidimensional integrodifferential equations for intramolecular nuclear motion, using Rayleigh–Schrödinger perturbation theory (RSPT), is tedious and error prone. For example, a 4th-order perturbation treatment for a triatomic molecule results in ~500 of pages of equations [4]. In addition, the manual solution of the molecule results in ~500 of pages of equations. Second-order perturbation equations for transition intensities lead to ~100 pages of equations for just triatomic molecules [5]. These algebraic solutions for the RSPT equations are found using symbolic mathematics and automatically translated into FortranForm via Mathematica. The subsequent calculation of molecular energies, intensities and properties scales linearly with the order of RSPT [6].
The second is the analytical representation of numerical PESs, where scaling issues seen in first-principles quantum mechanical perturbation theory treatments of intramolecular motion are traced to analytical representations of PESs, accomplished here using singular value decomposition (SVD) [7] For example, using SVD, a 6th-degree power series expansion of the PES for a 12-atom molecule, having 30 vibrational degrees of freedom, requires the inversion of rank 2-million matrix [2]. However, inversions of high-rank matrices also scale linearly and are accomplished straightforwardly and efficiently using modern parallel supercomputers [8].
In order to describe vibrational and rotational states, the Schrödinger equation for intramolecular nuclear motion is solved in the context of the Born–Oppenheimer approximation. The equation can be cast into a form involving analytic forms for the potential energy operator V in the Hamiltonian operator. This was accomplished for the ground vibrational state of triatomic molecules by Kern and Matcha using variation perturbation theory [9]. It has been solved for any polyatomic molecule using RSPT for the I = 1, (3N-6) modes of vibration in terms of normal coordinates Qi, as follows (in atomic units) [2]:
H = T + V
H = H0 + H1 + H2
H1 = ∑kijkqiqjqk = V1
In Equations (1)–(5) T is the total kinetic energy operator defined in terms of the harmonic frequencies ωi, the mass-reduced normal coordinates qi = Qi/√ωi and the momenta pI = −i ∂/∂qi. The second term in Equation (5) corresponds to the correction due to Coriolis interactions. Iα are the moments of inertia relative to the principal moment of inertia axes α = A, B, and C, and Πα are the vibrational total angular momenta.
Normal coordinates can be obtained using a finite differences procedure for calculating the second derivatives of the total energy in terms of mass-weighted cartesian nuclear displacement coordinates, the Hessian matrix. The eigenvectors (Lij) of the Hessian matrix comprise the linear transformation matrix that defines the normal coordinates in terms of the mass weighted cartesian displacement coordinates. The eigenvalues of the Hessian matrix are the harmonic frequencies [2]. The constants kijk, and kijkl are cubic and quartic force constants that account for anharmonicity. These constants are calculated from an initial power series fit of the PES in terms of internal coordinates by means of SVD, using the normal coordinate transformation matrix Lij [2]. The total potential energy operator to the second-order of RSPT is then
V = V0 + V1 + V2 + V2′
Solution of the RSPT equations leads to the following expression for vibrational term energies, the anharmonicity constants being algebraic formulae that only involve the ωi. kijk, and kijkl constants, obtained as noted above. Any desired line in the vibrational spectrum can be calculated from term energy differences.
Equation (7) defines the vibrational energy for any asymmetric top (a) molecule (moments of inertia Ae ≠ Be ≠ Ce) expressed in the terms of harmonic (ωi) and anharmonic (ωij) spectroscopic constants.
An analogous derivation, using RSPT the energy levels of a symmetric top (s) molecule (Ae = Be ≠ Ce or Ae ≠ Be = Ce), results in the term energy formula [2].
In Equation (8) the subscripts r and s refer to nondegenerate modes of vibration and t and t’ refer to degenerate ones. The so-called Dunham constant G in Equations (7) and (8) is an additive constant, also defined algebraically in terms of the ωi. kijk, and kijkl constants, that does not depend on vibrational or rotational quantum numbers [2].
Energies EvJ for a rotational level JvB within vibrational level v are given by
EvJ = Bv JvB (JvB + 1)
For an asymmetric top molecule, the rotational constant along the principal axis B is Bav = Beq − ∑r (vr + ).
For a symmetric top molecule, the rotational constant along the principal axis B is
where the rotational constants Bv are defined in terms of vibration-rotation coupling constants αB, the doubling constant qt and the vibrational angular momentum quantum number lt. [2] Equations defining rotational levels JvA and JvC with respect to the A and C principal moment of inertia axes are parallel to Equations (9) and (10).
Finally, the term energy T (v,J) for a state defined by vibrational quantum numbers vi, i = 1, (3N-6) and corresponding rotational quantum numbers Jv is given by
T (v,J) = Ev + EvJ
Thus, the frequency v of any spectral line corresponding to a change v’ to v in vibrational and J’ to J rotational sets of quantum numbers can be calculated as a term energy difference by using Equation (11), that is:
v = T (v’,J’) – T (v,J)
It is noted that the constant G, also known the lead Dunham constant Y00, in Equations (7) and (8) cancels when Equation (12) is used to calculate a line frequency, since it does not depend on vibrational or rotational quantum numbers.
Based on the RSPT formalism outlined above, a general-purpose code Survibtm has been published for the vibrational-rotational analysis of polyatomic molecules [6]. Survibtm was developed for fitting multi-dimensional potential energy and property surfaces and calculating vibrational-rotational spectra, line intensities and property expectation values of symmetric and asymmetric top polyatomic molecules. The code performs seven main types of calculations, all based on providing PESs and corresponding surfaces for molecular properties. These include (1) determining the equilibrium (stationary) point and saddle point geometries; (2) calculating internal coordinate force fields based on power series expansions in internal coordinates; (3) performing normal mode analyses; (4) calculating spectroscopic constants based on RSPT to second order; (5) calculating expectation values of properties; and (6) calculating transition dipole moments and line intensities [3]. Survibtm is interfaced with MolecGeom and employed in this study to analyze a given PES. As noted above, the components of a PES include the internal coordinates (bond lengths, bond angles, dihedral angles) and the total energies of a molecule. Given a PES, Survibtm solves for spectroscopic constants which include anharmonicity through quartic terms according to second-order RSPT, as shown in Equations (7)–(10).
RSperturb and DiatomicVibRot are Mathematica codes that analyze high-order perturbation methods using symbolic mathematics [10]. Rsperturb provides algebraic solutions to the quantum mechanical molecular vibrational-rotational problems for a diatomic molecule, given its potential energy curve (PEC) (a PEC is the limiting case of a PES involving just two atoms). For example, differences observed between the theoretical sixth-order RSPT results and experimental energies for H2, N2, CO and HF spectral lines are typically less than 0.1%. Vibrational and rotational energies for H2, calculated using DiatomicVibRot, indicate how accuracy degrades slowly for higher-lying vibrational and rotational states, as is expected in perturbation theory [2]. It is noteworthy that current state-of-the-art solutions for polyatomic systems have been derived only up to the second order of perturbation theory for polyatomic molecules [11]. Expanding on previous published work’s theoretical developments will be necessary in order to calculate the arbitrary-order correction terms to vibrational-rotational energies that allow for accurate calculations of polyatomic vibrational-rotational spectra for spherical, symmetric, and asymmetric top molecules.
The algebraic equations resulting from high-order perturbation theory can be properly and efficiently treated to the appropriate level of approximation as dictated by the accuracy of Born–Oppenheimer PESs. The Born–Oppenheimer approximation is an assumption which states nuclear and electronic motion within molecules can be separated. In a follow-up study, the RSperturb and Survibtm codes will be combined, modified and extended to be applicable to polyatomic molecules. The derivation includes the terms in the Hamiltonian operator required to calculate, to arbitrary-order, anharmonicity effects, intensities, non-rigid rotations, vibration-rotation couplings, wave functions and property expectation values. The requisite correction formulae in terms of universal constants, molecular constants, and quantum numbers are to be generated using symbolic computation through a generalization of the q[n] procedural program [10] to polyatomic systems within Mathematica. The necessary equations, governing such high-order effects as resonances, Coriolis couplings and centrifugal distortions, are to be incorporated. The algebraic results will be translated into computer code at runtime using a supplied analytical representation of a molecular PES and will be executed on an advanced computing platform to calculate ab initio spectra and related properties [5].
When calculating spectra there are several different properties that can be compared with what is observed: e.g, fundamental transitions, hot bands, combinations and overtones. A fundamental transition is a symmetric stretch or bend, or asymmetric stretch corresponding to an excitation from the ground vibrational state to the first excited vibrational level in bent triatomic molecules. Hot bands are transitions from excited-to-excited vibrational states. Combinations are simultaneous excitations of multiple normal modes with a single photon. Overtones are also observed in IR spectroscopy and occur when a molecule undergoes a transition from the ground state to the second or higher excited state, meaning that the vibrational quantum number of the upper state is greater than one excitation. Consequently, through the execution of the MolecGeom code, a swift and reliable generation of spectra for polyatomic molecules is within reach.
2. Method
An accurate quantum mechanical analysis of intramolecular nuclear motion by means of PESs requires saturating the molecular geometry space in an efficient manner. The MolecGeom code presented here enables the generation of bond lengths, bond angles and dihedral angles for any polyatomic molecule. This code reduces the time necessary for the generation of extremely lengthy geometric output, while minimizing the error involved in the requisite SVD analysis. This is accomplished through a Mathematica-based algorithm that ensures an error-free process. The MolecGeom code for any polyatomic system is as follows:
Input 1:
MolecGeom = Flatten [Table [{BL 1, BL 2, BL 3…, ANG 1, ANG 2, ANG 3...},
{BL 1, RS, RE, INC}, {BL 2, RS, RE, INC}, {BL 3, RS, RE, INC},
{ANG 1, RS, RE, INC}, {ANG 2, RS, RE, INC}, {ANG 3, RS, RE, INC}], 2]
Output 1:
{BL 1+INC, BL 2+INC, ANG 1+INC, ANG 2+INC},
{BL 1+INC+INC, BL 2+INC+INC, ANG 1+INC+INC, ANG 2+INC +INC}
Here, MolecGeom is a variable that specifies the intramolecular nuclear positions of any polyatomic molecule. Furthermore, BL 1, BL 2, BL 3 …, are the terms used to describe reference geometry bond lengths, while ANG 1, ANG 2, ANG 3..., are the variables used to describe the reference geometry bond angles for any polyatomic molecule. The following input, BL 1, RS, RE, INC, where BL 1, the reference geometry bond length 1 from the polyatomic molecule in question, is placed in this slot. RS translates to range start, meaning the value of the increment above the reference geometry bond length where the iterations begin, while RE is defined as range end, meaning the value of the increment below the reference geometry bond length where the iterations end. Lastly, INC, is defined as increment, meaning the number of times the specified RS and RE should step up or down and calculate the corresponding geometric parameters. Lastly, the output produced is formatted as shown above in Output 1.
Following the calculation, another short series of steps is completed to ensure all points produced are unique with respect to the molecular point group symmetry. This is accomplished by querying Mathematica for the dimensions of the geometric output. These dimensions are then multiplied and placed into a variable x. This variable is then used to reshape the array, and then any duplicate points are deleted. This finalized set is then exported into a text file for further implementation. Below are input examples used to finalize and prepare the geometric output for further implementation.
Input 2: MolecGeom//Dimensions
This will provide the dimensions of Output 1. Using formaldehyde as an example, the output would be the following:
Output 2: {27,4,4,5,6}
These values 27, 4, 4, and 5 are the dimensions of the data set produced from Input 1, corresponding to the 6 internal coordinates of formaldehyde  : two CH bond lengths, CO bond length, two HCO bond angles, and the H2C dihedral wagging angle.
: two CH bond lengths, CO bond length, two HCO bond angles, and the H2C dihedral wagging angle.
: two CH bond lengths, CO bond length, two HCO bond angles, and the H2C dihedral wagging angle. Input 3:
x = 27 × 4 × 4 × 5
Output 3:
2160
Allowing the numbers to be stored as a single variable will allow for the array reshape to be completed.
Input 4: Data = ArrayReshape[MolecGeom, {x, 6}]
Output 4:
{2.2, 2., 2., 115, 115, 170}
{2.2, 2., 2., 115, 115, 175}
{2.2, 2., 2., 115, 115, 180}
{2.2, 2., 2., 115, 115, 185}
{2.2, 2., 2., 115, 115, 190}
{2.2, 2., 2., 115, 120, 170}
{2.2, 2., 2., 115, 120, 175}
{2.2, 2., 2., 115, 120, 180}
{2.2, 2., 2., 115, 120, 185}
{2.2, 2., 2., 115, 120, 190}
The new name “Data” will hold the MolecGeom dataset by reformatting the numbers in {x,6}; that is, {B1, B2, B3, A1, A2, A3}, etc.
Input 5:
Data//DeleteDuplicates//Dimensions
Output 5:
{2160,6}
The following input will allow for the formatted data set “Data” to be scanned via Mathematica to remove any symmetry replicated points (DeleteDuplicates). When generating PESs, this is useful to avoid infinite loops and errors of non-convergence.
Input 6:
Export[“Data. Txt”, Data]
Output 6:
Data.txt
Lastly, in order to export data effectively, the “Export” command is used to move the formatted set “Data” out of the Mathematica Wolfram notebook. This file is then deposited on the local computer. The final internal coordinate dataset is then ported to a compute engine to calculate energies that define PESs for vibrational rotational analysis [6].
3. Application
Example calculations have been performed by generating multiple internal coordinate geometric data sets for four molecules, water (H2O) [12], formaldehyde (H2CO) [13,14], vinyl alcohol (C2H4O)[15,16] and vitamin C [ascorbic acid (C6H8O6)] [17,18]. By implementing MolecGeom PES sizes ranging from as few as 200 to more than 20,000 geometries have been defined. In order to generate molecule-specific geometric output, it is imperative to note that reference bond lengths and bond angles must be specified. In the case of water, these correspond to the equilibrium bond lengths (OH1 and OH2) of 1.81 atomic units (au; 1 au = 0.529177 Å) and the equilibrium bond angle (HOH) of 104.5°. These are used as a starting point to select a range of geometric distortions of 0.005 au, above and below the range start of 1.80 au and the range end of 1.82 au. The angle of the range start is 103.5° and the range end is at 105.5°, and increments are calculated every 0.100° in the vicinity of equilibrium values. Applying these parameters for water, the MolecGeom code generates a 525-geometry surface.
In the case of formaldehyde, the equilibrium CO, CH1, and CH2 bond lengths are 2.30, 2.10, and 2.10 au, along with two HCO and the H2C dihedral wagging bond angles of 120.0°, 120.0° and 180.0°. These are then used as a starting point to select a range of numbers that lie around these equilibrium values. The range start for the CO calculation is 2.20 au and the range end is 2.40 au, with an incrementation of 0.09 au. The range start for the first and second CH stretch is 2.00 au and the range end is set at 2.20 au, with the same 0.09 au incrementation. Next, the distortions of the two HCO angles were calculated. Their range start begins at 115.0° and the range ends at 130.0°, with 5.0° increments. The wagging angle range start begins at 170.0° and the range end is set to 190.0°, with 5.0° incrementations. The MolecGeom code calculated a new geometry with every incrementation within each range start and end, thus creating a comprehensive geometric list for formaldehyde containing 2160 geometries for follow-on implementation.
After importing the respective data sets to a compute engine, calculations are carried out to generate the energies corresponding to these PESs of water and formaldehyde. That is, these calculations produce energies for all the MolecGeom-generated geometric points for each of these polyatomic molecules. These PESs are then ported into a modified version of Survibtm [6] to calculate the fundamental vibrational frequencies shown in Table 1 and Table 2 below. For reference, the fundamental visual vibrations of water and formaldehyde are shown in Figure 1 and Figure 2, respectively.

Table 1.
Water fundamental vibrational frequencies (cm−1) from a 125-geometry surface with comparisons with observed values [19].
Table 2.
Formaldehyde fundamental frequencies (cm−1) calculated from a 2160-geometry surface with comparisons with observed values [19].
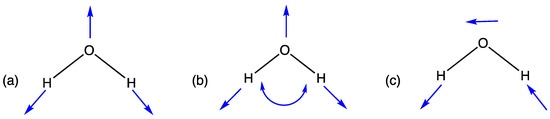
Figure 1.
Visual vibrations of water (a) O-H symmetric stretch, (b) H-O-H bend and (c) O-H asymmetric stretch.
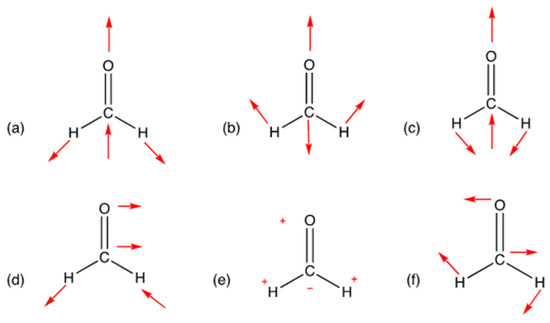
Figure 2.
The six fundamental transitions corresponding to formaldehyde are depicted. (a) H-C-H Symmetric stretch (b) C=O stretch (c) H-C-H Scissoring (d) H-C-H Asymmetric stretch (e) CH2 out of plane wagging (f) H-C-H Rock.
In Table 1, the calculated value of 3654 cm−1 denotes the symmetric stretch of the water molecule, meaning the O-H bonds are stretched equally. The following value of 1595 cm−1 corresponds to the bending motion that occurs where the water molecule bond angle opens and closes. The last value of 3753 cm−1 describes the asymmetric stretching of the molecule, where one O-H bond is contracted and the other O-H bond is stretched and vice versa.
Furthermore, Table 2 contains the six fundamental transition frequencies corresponding to the motions depicted in Figure 2 for formaldehyde, as shown above. These are calculated using a PES comprised of 2160 geometries generated using MolecGeom. In Table 2, the calculated value of 2803 cm−1 corresponds to CH2 symmetric stretch, meaning the H-C-H bonds are stretched equally. The following value reported of 1749 cm−1 corresponds to C=O stretch, meaning the double bond is stretched upwards. In continuation, 1502 cm−1 corresponds to the CH2 scissoring; this is the movement of two atoms toward and away from each other. Furthermore, the 2819 cm−1 value reported resembles the CH2 asymmetric stretching, where the C-H bond is contracted, and the other C-H bond is stretched and vice versa. Additionally, 1168 cm−1 relates to the CH2 wagging motion; this motion can be described as a “v” shape moving side to side. Lastly, 1246 cm−1 corresponds to the CH2 rocking; this motion can be pictured as a “v” opening and closing.
Following the analysis of the vibrational-rotational transitions, quantum mechanical averages (expectation values) of molecular properties, such as dipole moments and electric field gradients, can also be calculated using Survibtm, as well as other codes such as SPECTRO [20]. These are based on surfaces for these properties analogous to the PESs. Additionally, transition intensities among vibrational levels can be calculated using dipole moment surfaces [21].
Lastly, in order to show the robustness of MolecGeom for larger molecules, vinyl alcohol and ascorbic acid (vitamin C) were selected as examples. In the case of vinyl alcohol, shown in Figure 3, there are six equilibrium bond lengths (B), along with five bond angles(A) and three dihedral angles (D) for this molecule. More specifically: B1 = 1.07 Å, B2 = 1.07 Å, B3 = 1.35 Å, B4 = 1.07 Å, B5 = 1.43 Å, B6 = 0.96 Å, A1 = 120.0°, A2 = 120.0°, A3 = 120.0°, A4 = 120°, A5 = 109.0°, D1 = 180.0°, D2 = 180.0°, and D3 = 150.0°. These equilibrium values were generated using Gaussview 16; the structure can be seen below [22].
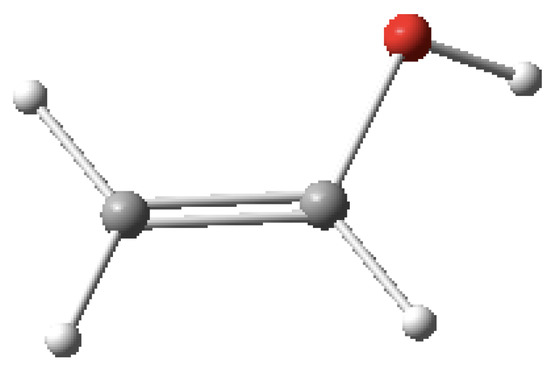
Figure 3.
Vinyl alcohol (C2H4O) image generated in Gaussview 16 [22].
The equilibrium values generated are used as a starting point to select a range of geometric distortions of 0.20 Å above and below the range start of 1.00 Å and the range end of 1.10 Å, for B1, B2 and B4. The range start for B3 is 1.34 Å and the range end is set at 1.37 Å with the same 0.20 Å incrementation. Next, the distortions of the five angles were calculated. The range start begins at 115.0° and the range ends at 120.0°, with 5.0° increments for A1, A2, A3 and A4. For the last distortion, A5, the range start begins at 106.0° and the range ends at 112.0°, with 5.0° increments. The MolecGeom code calculated a new geometry at every incrementation within each range start and end, thus creating a comprehensive geometric list for vinyl alcohol, containing 1458 unique geometries for follow-on implementation into Survibtm for the calculation of its vibrational-rotational spectrum.
In the final example, the ascorbic acid (C6H8O6) molecule is composed of 20 atoms with 19 bond lengths, 18 bond angles and 17 dihedral angles for a total of 54 internal coordinates. Shown in Table 3 are the internal coordinates that were inputted to MolecGeom. A total of 1,899,776 geometric points were calculated, which define its PES. More specifically, the equilibrium values seen in Table 3 were generated using Gaussview 16. The structure is shown in Figure 4 [22].
Table 3.
Ascorbic acid molecular geometry distortions with range start (RS), range end (RE) and increments (INC). Where B1–B19 are in angstroms and A1–D17 are in degrees.
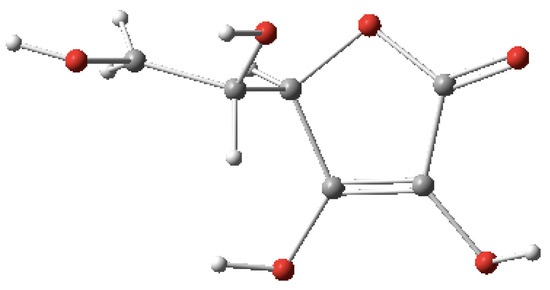
Figure 4.
Ascorbic acid (C6H8O6) image generated in Gaussview 16 [22].
File S1: The notebook (.nb) files for water, formaldehyde, vinyl alcohol and ascorbic acid have been provided. Additionally, File S2: The text (.txt) files containing the unique geometric points for each molecule are also provided. These can be accessed through the link in the Supplementary Materials section.
4. Conclusions
The MolecGeom code reported here allows for the rapid and efficient generation of geometric data for PESs. Mathematica provides an ideal programming environment for addressing the algebraic manipulations required to define such PESs. Additionally, depending on the required application of the PES generated, the parameters of MolecGeom can be adjusted as needed. In this study, the code is applied to water, formaldehyde, vinyl alcohol and ascorbic acid molecules; however, it can be used to generate a PES for any polyatomic system, while also recognizing the molecular point group symmetry relations. By applying the results of MolecGeom, accurate vibrational-rotational spectra are attainable. The errors in the calculations of fundamental frequencies for water and formaldehyde are shown to be less than 1% in comparison with the published literature values for PESs, based on accurate ab initio electronic energy and properties calculations. The MolecGeom code provides an effective means by which accurate PESs for vibrational analyses of polyatomic molecules may be realized.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/a16010006/s1, File S1: The notebook (.nb) files for water, formaldehyde, vinyl alcohol and ascorbic acid have been provided. Additionally, File S2: The text (.txt) files containing the unique geometric points for each molecule are also provided.
Author Contributions
Conceptualization, A.R.C. and W.C.E.; methodology, A.R.C. and W.C.E.; software, A.R.C. and W.C.E.; validation, A.R.C. and W.C.E.; formal analysis, A.R.C. and W.C.E.; investigation, A.R.C. and W.C.E.; resources, A.R.C. and W.C.E.; data curation, A.R.C. and W.C.E.; writing—original draft preparation, A.R.C. and W.C.E.; writing—review and editing, A.R.C. and W.C.E.; visualization, A.R.C. and W.C.E.; supervision, W.C.E.; project administration, W.C.E.; funding acquisition, A.R.C. and W.C.E. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by the RISE PhD program at UTSA funded by NIH/NIGMS RISE GM60655. Additionally, this research was supported in part by the National Science Foundation under grant CHE-1853477.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Data available in a publicly accessible repository. The data presented in this study are openly available at [https://www.mdpi.com/article/10.3390/a16010006/s1].
Acknowledgments
This work received computational support from UTSA’s HPC cluster Arc, operated by the university technology solutions research computing support group. Lastly, we are grateful to Wolfram Technology Support Services for their troubleshooting help.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Collins, M.A. Molecular potential-energy surfaces for chemical reaction dynamics. Theor. Chem. Acc. 2002, 108, 313–324. [Google Scholar] [CrossRef]
- Ermler, W.C.; Hsieh, H.C. Analytical Representaiton and vibrational-rotational analysis of AB Initio potential energy and property surfaces. Adv. Mol. Electron. Struct. Theory 1990, 1, 1–44. [Google Scholar]
- Wolfram Research, Inc. Mathematica; Wolfram Research, Inc.: Champaign, IL, USA, 2022. [Google Scholar]
- Krohn, B.J. Calculation of Vibration-Rotation Energies of Diatomic Molecules to Sixth Order of Approximation and Bent Symmetrical XY2 Triatomic Molecules to Fourth Order by an Improved Systematic Perturbation Theory Procedure. Ph.D. Thesis, Ohio State University, Columbus, OH, USA, 1971. [Google Scholar]
- Krohn, B.J.; Kern, C.W. Nuclear corrections to molecular properties. VI. Vibrational transition moments in asymmetric-top molecules. J. Chem. Phys. 1978, 69, 5310–5313. [Google Scholar] [CrossRef]
- Ermler, W.C.; Hsieh, H.C.; Harding, L.B. Polyatomic surface fitting, vibrationalrotational analysis, expectation value and intensity program. Comput. Phys. Commun. 1988, 51, 257–284. [Google Scholar] [CrossRef]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vetterling, W.T. Numerical Recipes; Cambridge University Press: New York, NY, USA, 1986. [Google Scholar]
- Sherrill, C.D.; Schaefer, H.F., III. The Configuration Interaction Method: Advances in Highly Correlated Approaches. Adv. Quantum Chem. 1999, 34, 143–269. [Google Scholar]
- Kern, C.W.; Matcha, R.L. Nuclear Corrections to Electronic Expectation Values: Zero-Point Vibrational Effects in the Water Molecule. J. Chem. Phys. 1968, 49, 2081. [Google Scholar] [CrossRef]
- Herbert, J.M.; Ermler, W.C. Symbolic Implementation of Arbitrary order Pertubation Theory using Computer Algerbra: Application to Vibrational-Rotational Analysis of Diatomic Molecules. Comput. Chem. 1998, 22, 169–184. [Google Scholar] [CrossRef]
- Zhang, Q.; Day, P.N.; Truhlar, D.G. The accuracy of second order perturbation theory for multiply excited vibrational energy levels and partition functions for a symmetric top molecular Ion. J. Chem. Phys. 1998, 98, 4948. [Google Scholar] [CrossRef]
- Partridge, H.; Schwenke, D.W. The determination of an accurate isotope dependent potential energy surface for water from extensive ab initio calculations and experimental data. J. Chem. Phys. 1997, 106, 4618. [Google Scholar] [CrossRef]
- Yachmenev, A.; Yurchenko, S.N.; Jensen, P.; Thiel, W. A new “spectroscopic” potential energy surface for formaldehyde in its ground electronic state. J. Chem. Phys. 2011, 134, 244307. [Google Scholar] [CrossRef] [PubMed]
- Feller, D.; Dupuis, M.; Garrett, B.C. Barrier for the H2CO→H2+CO reaction: A discrepancy between high-level electronic structure calculations and experiment. J. Chem. Phys. 2000, 113, 218. [Google Scholar] [CrossRef]
- Bouma, W.J.; Radom, L. The structure of vinyl alcohol. J. Mol. Struct. 1978, 43, 267–271. [Google Scholar] [CrossRef]
- Peeters, J.; Nguyen, V.S.; Müller, J.F. Atmospheric Vinyl Alcohol to Acetaldehyde Tautomerization Revisited. J. Mol. Struct. 2015, 6, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibia, J.S.; Al Hazmib, Y.M.; Masmalib, K.M.; Gogary, T.M.E. Geometry and electronic properties of Vitamin C. Turkish Comp. Theo. Chem. 2018, 2, 39–44. [Google Scholar]
- Cozzolino, D.; Phan, A.D.T.; Netzel, M.E.; Smyth, H.; Sultanbawa, Y. The use of vibrational spectroscopy to predict Vitamin C in Kakadu plum powders. J. Sci. Food Agric. 2020, 101, 3208–3213. [Google Scholar]
- Shimanouchi, T. Tables of Molecular Vibrational Frequencies Consolidated. Natl. Bur. Stand. 1972, 1, 1–160. [Google Scholar]
- Willets, A.; Gaw, J.F.; Greeen, W.H.; Handy, N.C. SPECTRO, a Theoretical Spectroscopy Package; Bowman, J.M., Ed.; Advances in Molecular Vibrations and Collision Dynamics: Cambridge, UK, 1990. [Google Scholar]
- McCoy, A.B.; Boyer, M.A. Exploring Expansions of the Potential and Dipole Surfaces Used for Vibrational Perturbation Theory. J. Phys. Chem. A 2022, 126, 7242–7249. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision 6.0.16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).