
Fast and Interactive Positioning of Proteins within Membranes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
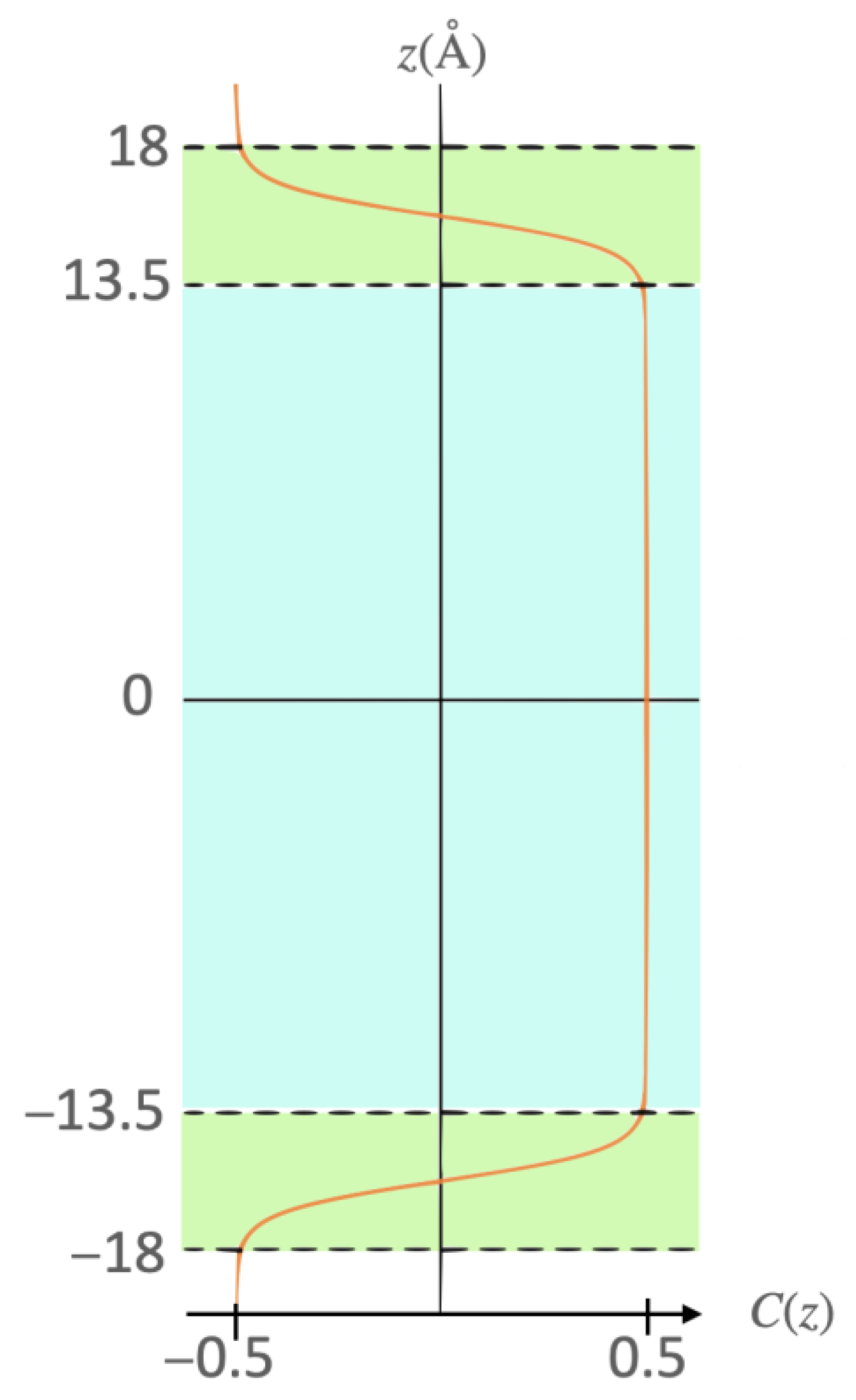
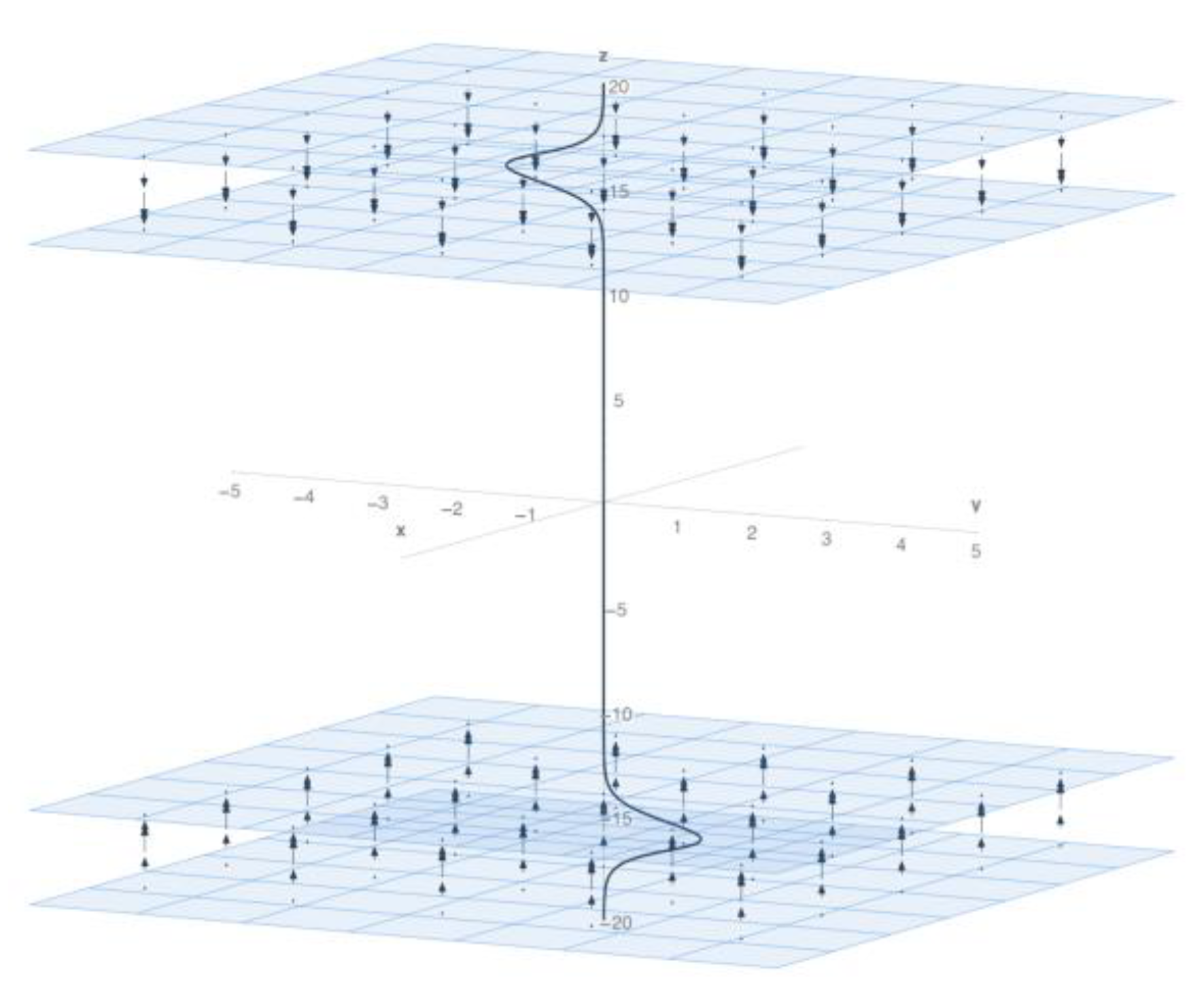
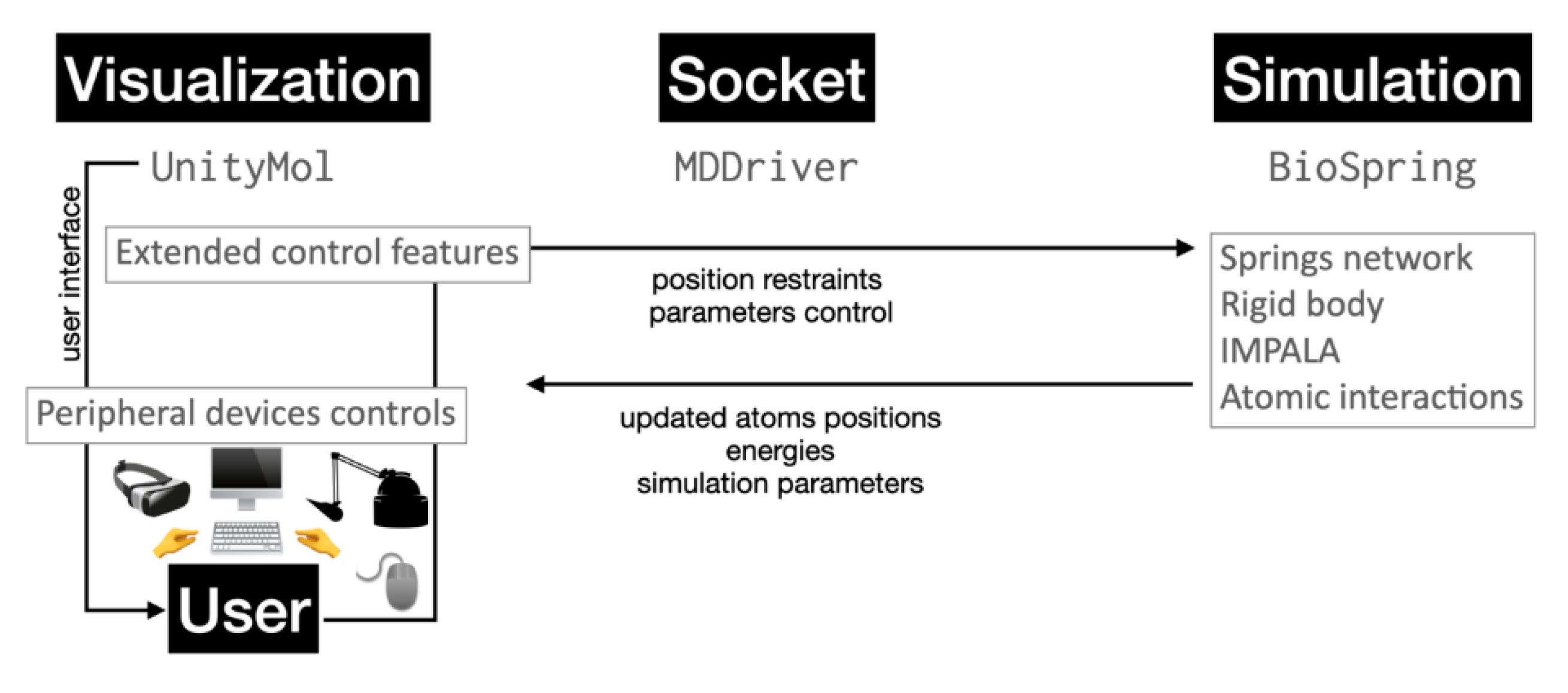
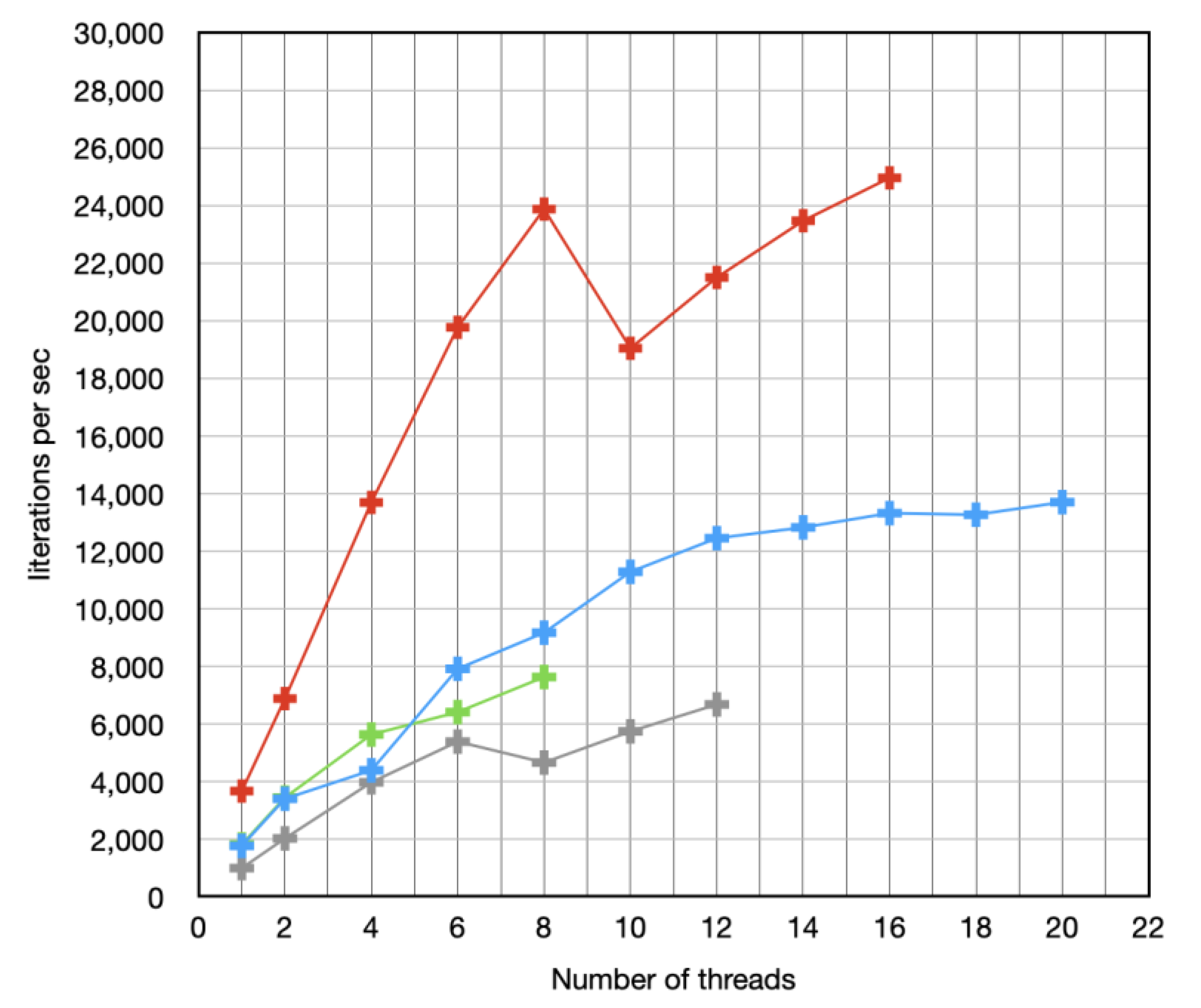
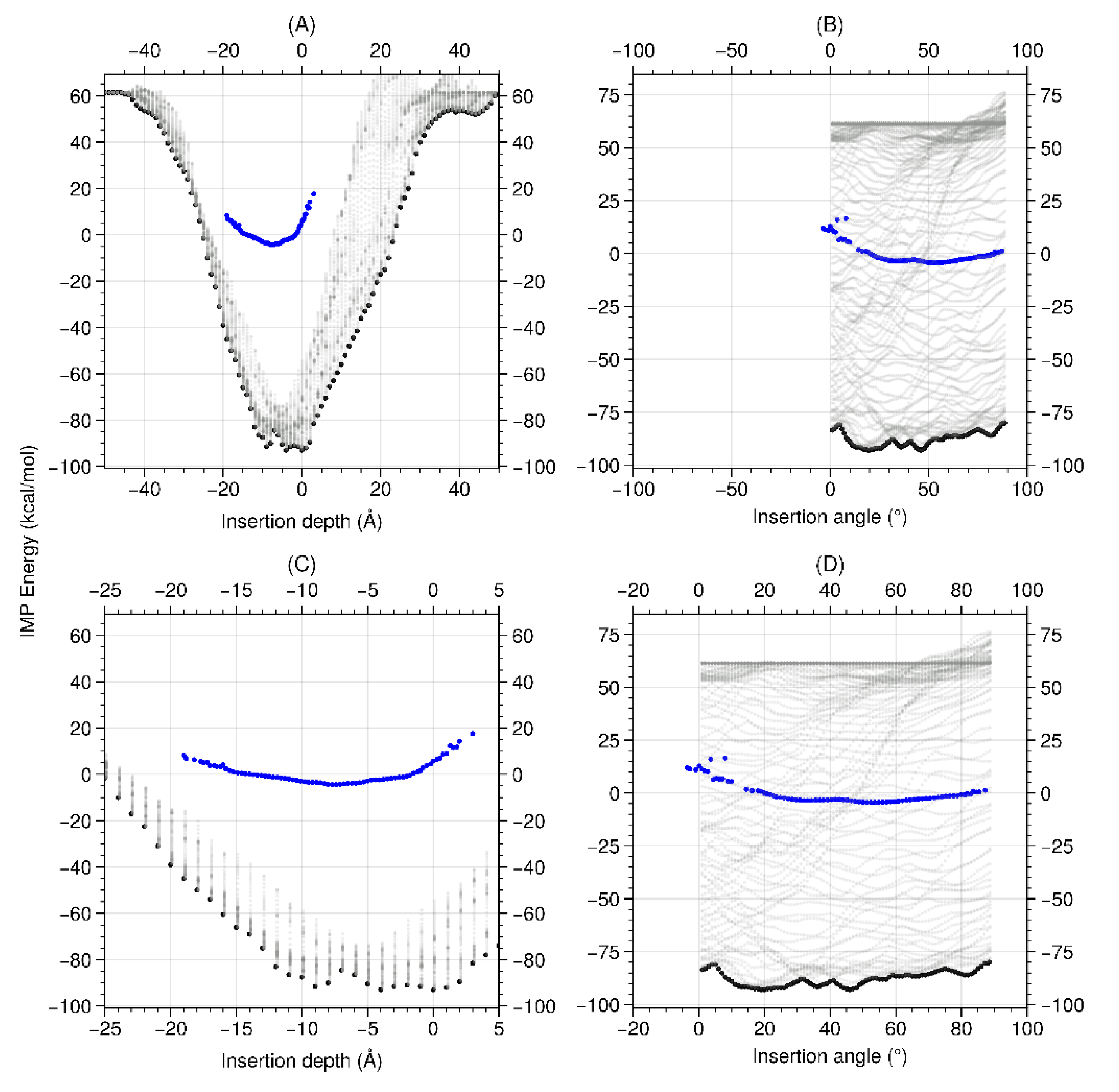
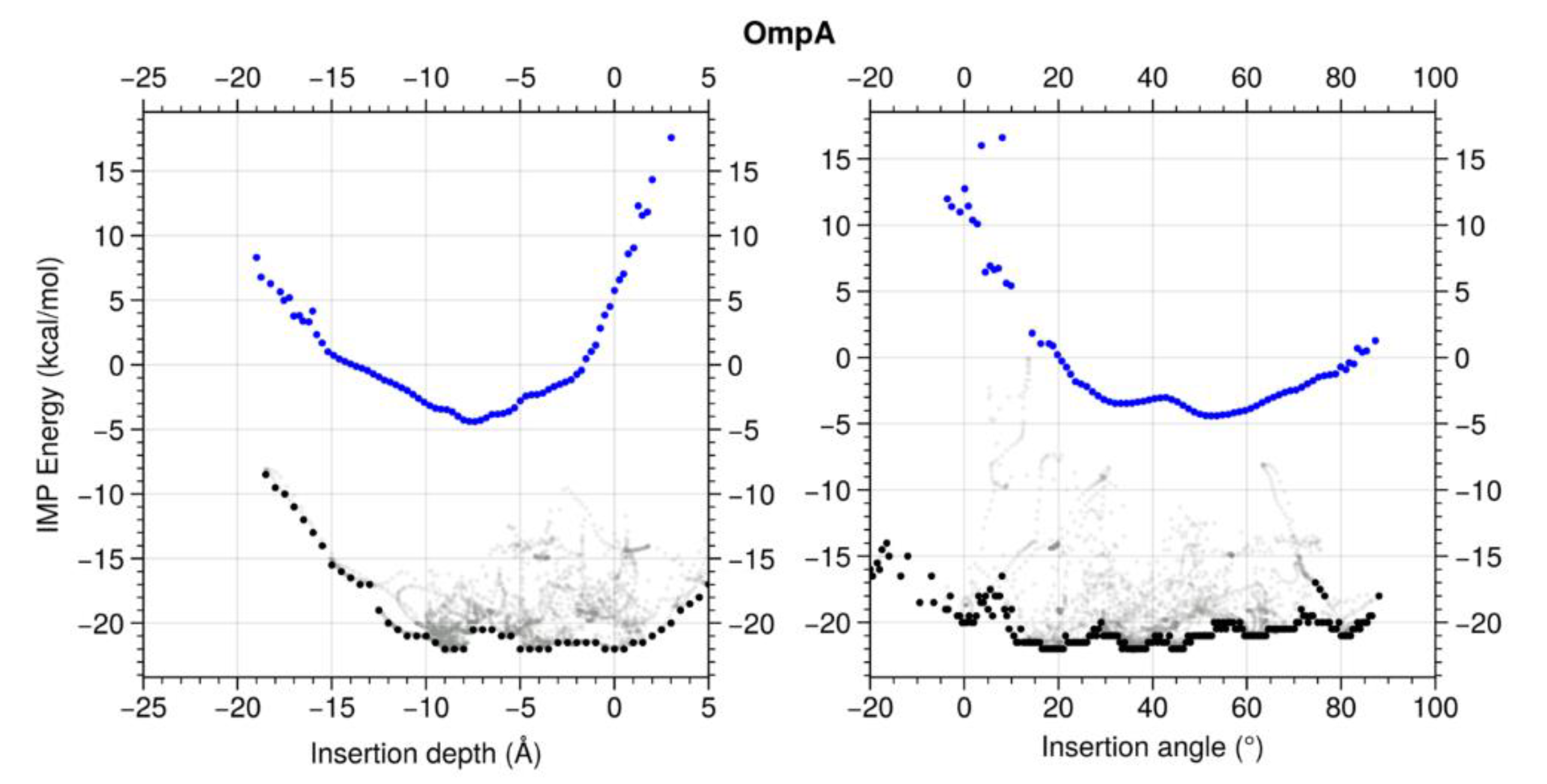
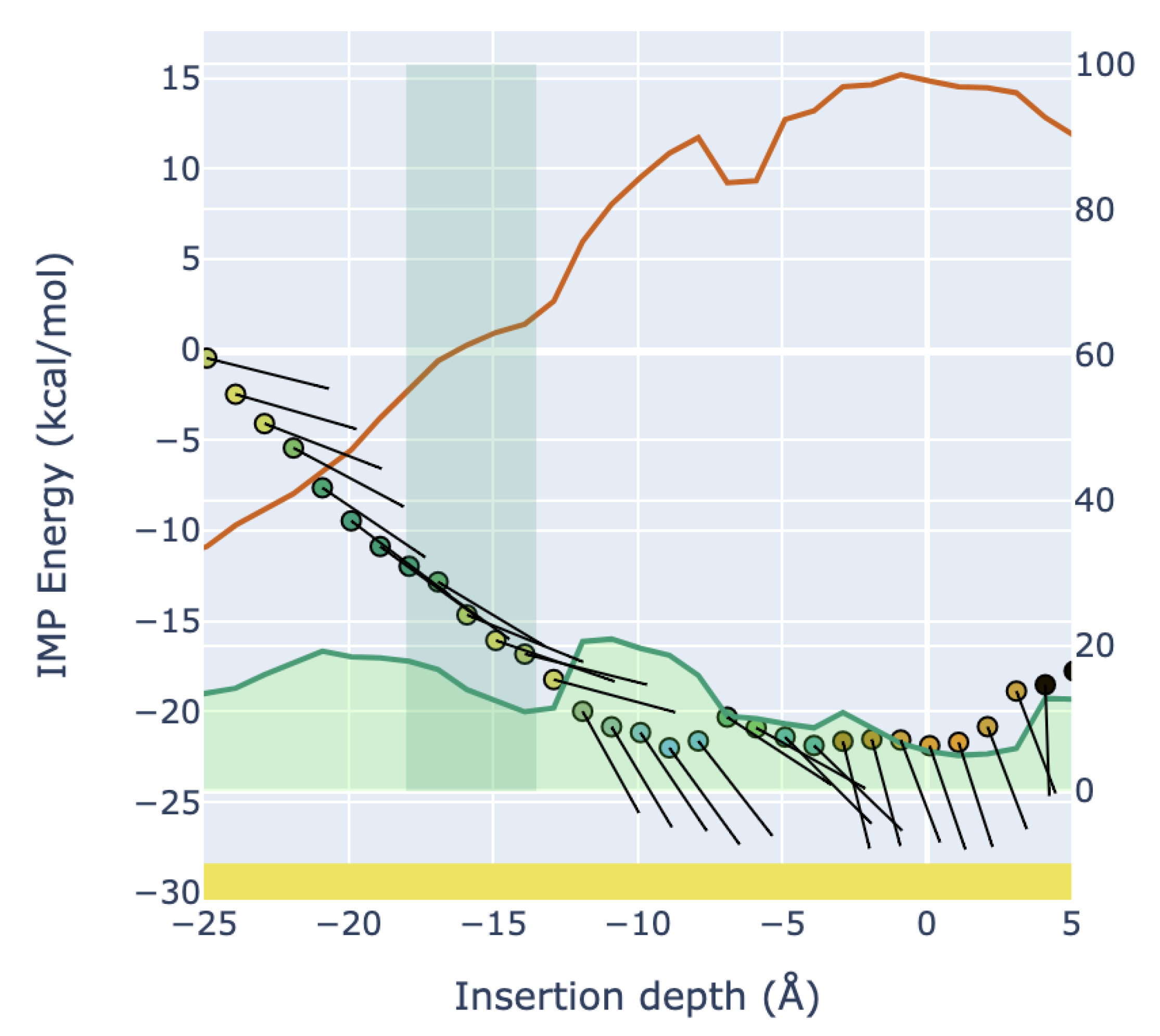
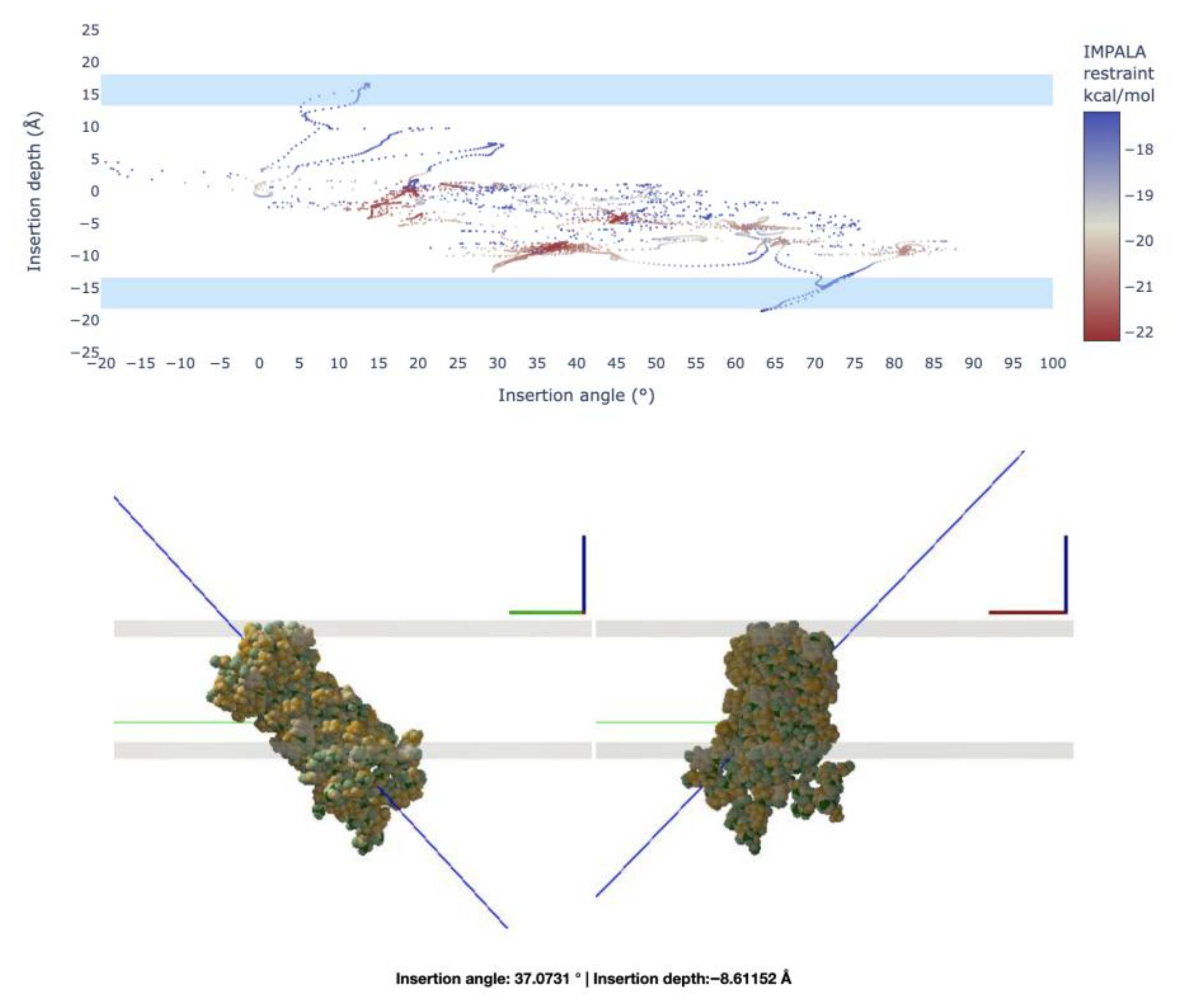
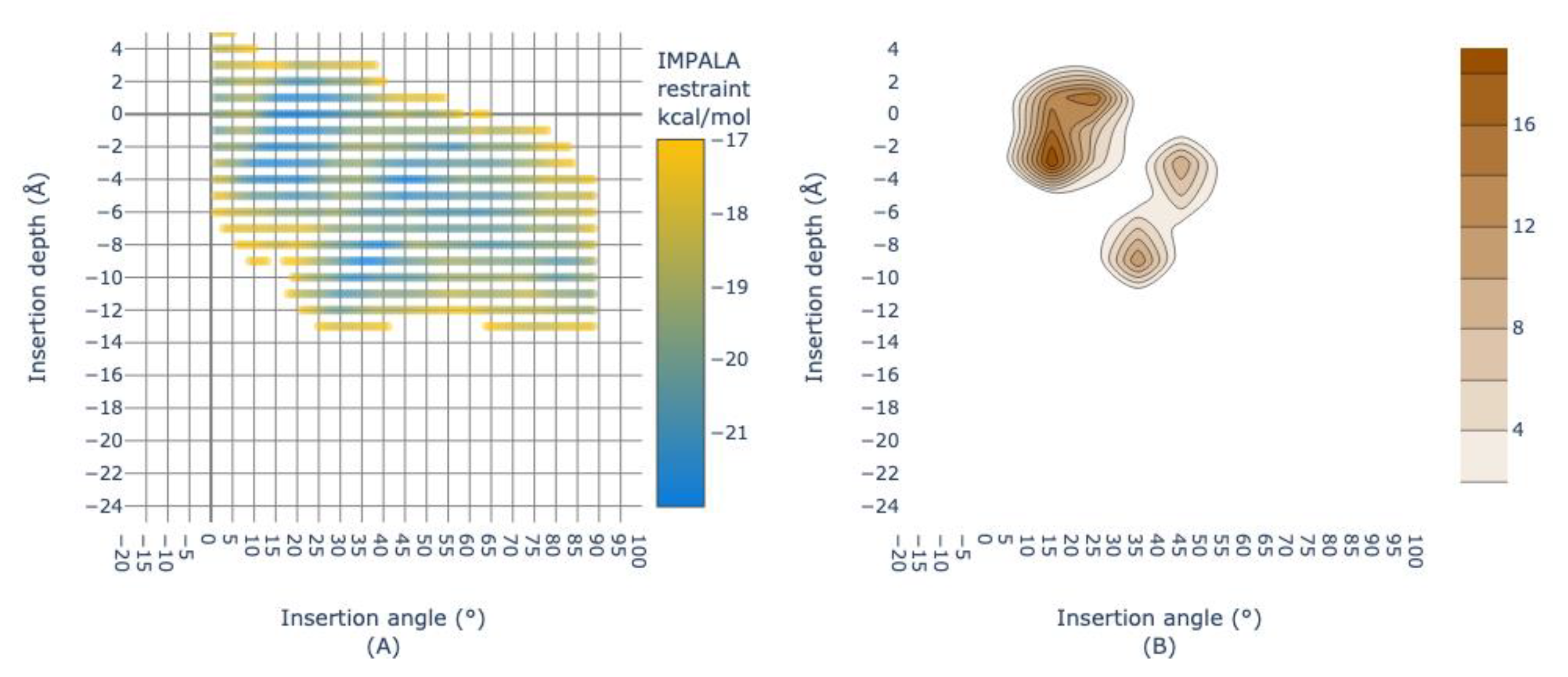
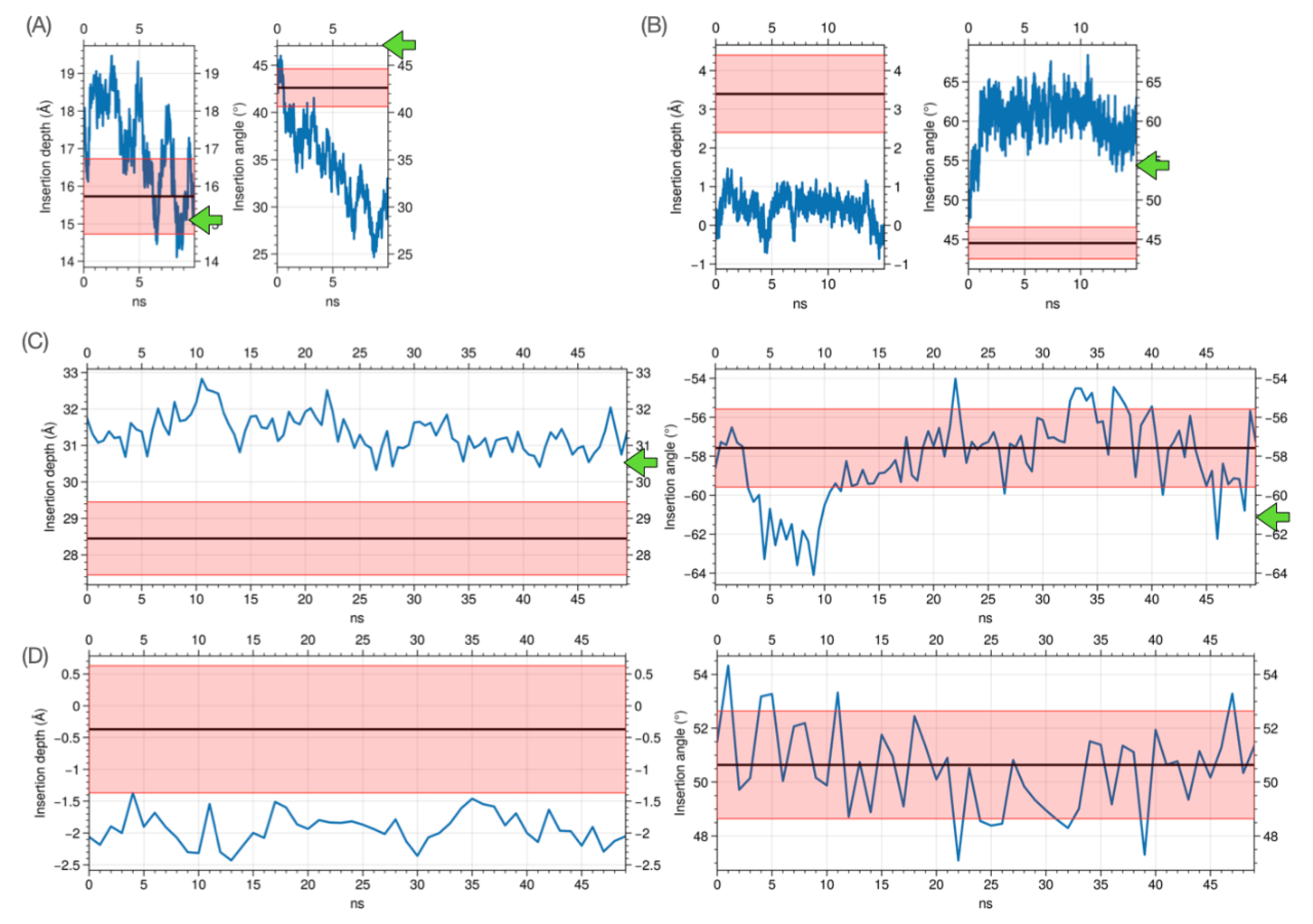
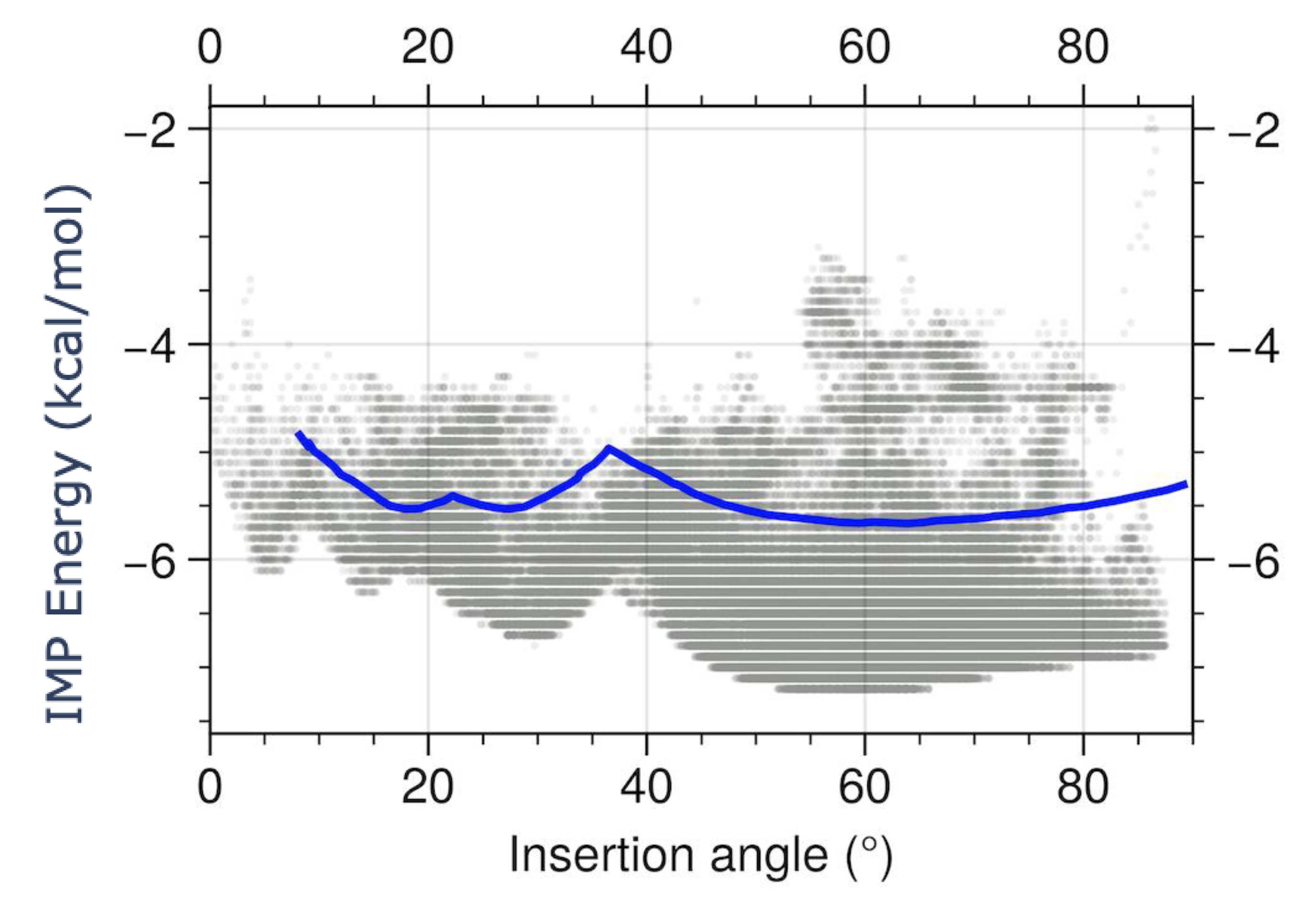
Abstract
Share and Cite
Lanrezac, A.; Laurent, B.; Santuz, H.; Férey, N.; Baaden, M. Fast and Interactive Positioning of Proteins within Membranes. Algorithms 2022, 15, 415. https://doi.org/10.3390/a15110415
Lanrezac A, Laurent B, Santuz H, Férey N, Baaden M. Fast and Interactive Positioning of Proteins within Membranes. Algorithms. 2022; 15(11):415. https://doi.org/10.3390/a15110415
Chicago/Turabian StyleLanrezac, André, Benoist Laurent, Hubert Santuz, Nicolas Férey, and Marc Baaden. 2022. "Fast and Interactive Positioning of Proteins within Membranes" Algorithms 15, no. 11: 415. https://doi.org/10.3390/a15110415
APA StyleLanrezac, A., Laurent, B., Santuz, H., Férey, N., & Baaden, M. (2022). Fast and Interactive Positioning of Proteins within Membranes. Algorithms, 15(11), 415. https://doi.org/10.3390/a15110415