
Role of Hydrogen Bonding in the Formation of Adenine Chains on Cu(110) Surfaces


Abstract
:1. Introduction
2. Results and Discussion
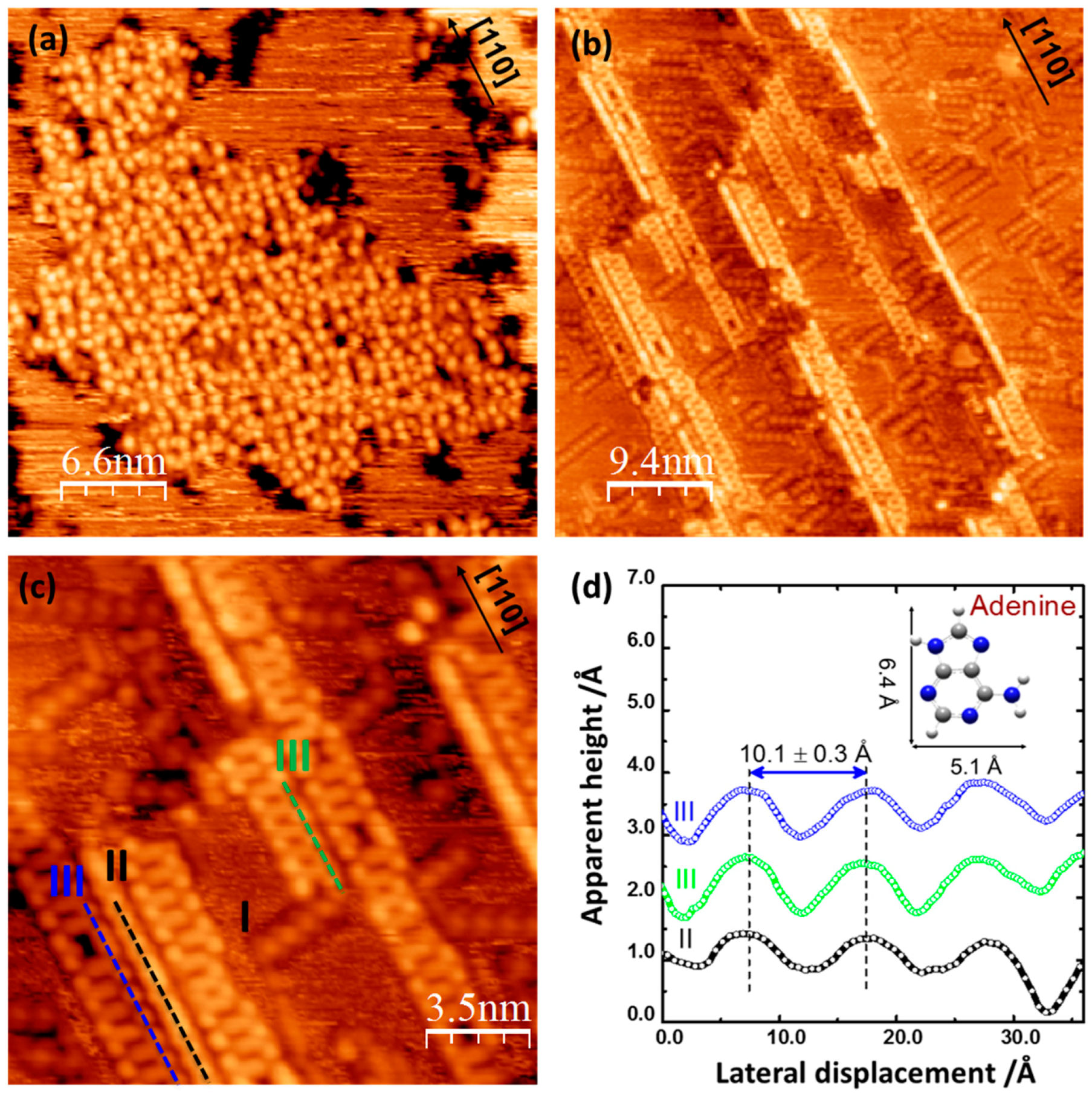
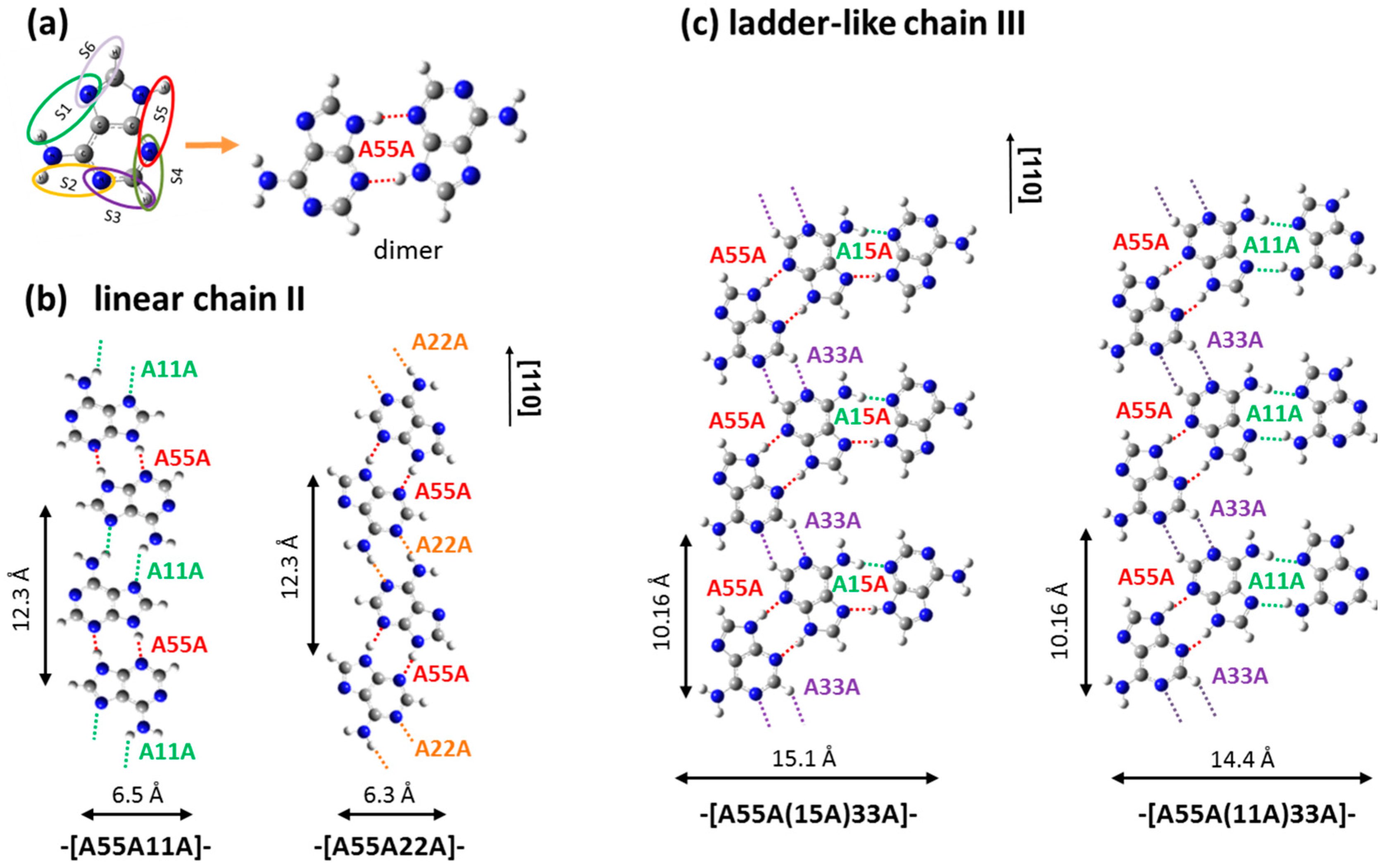
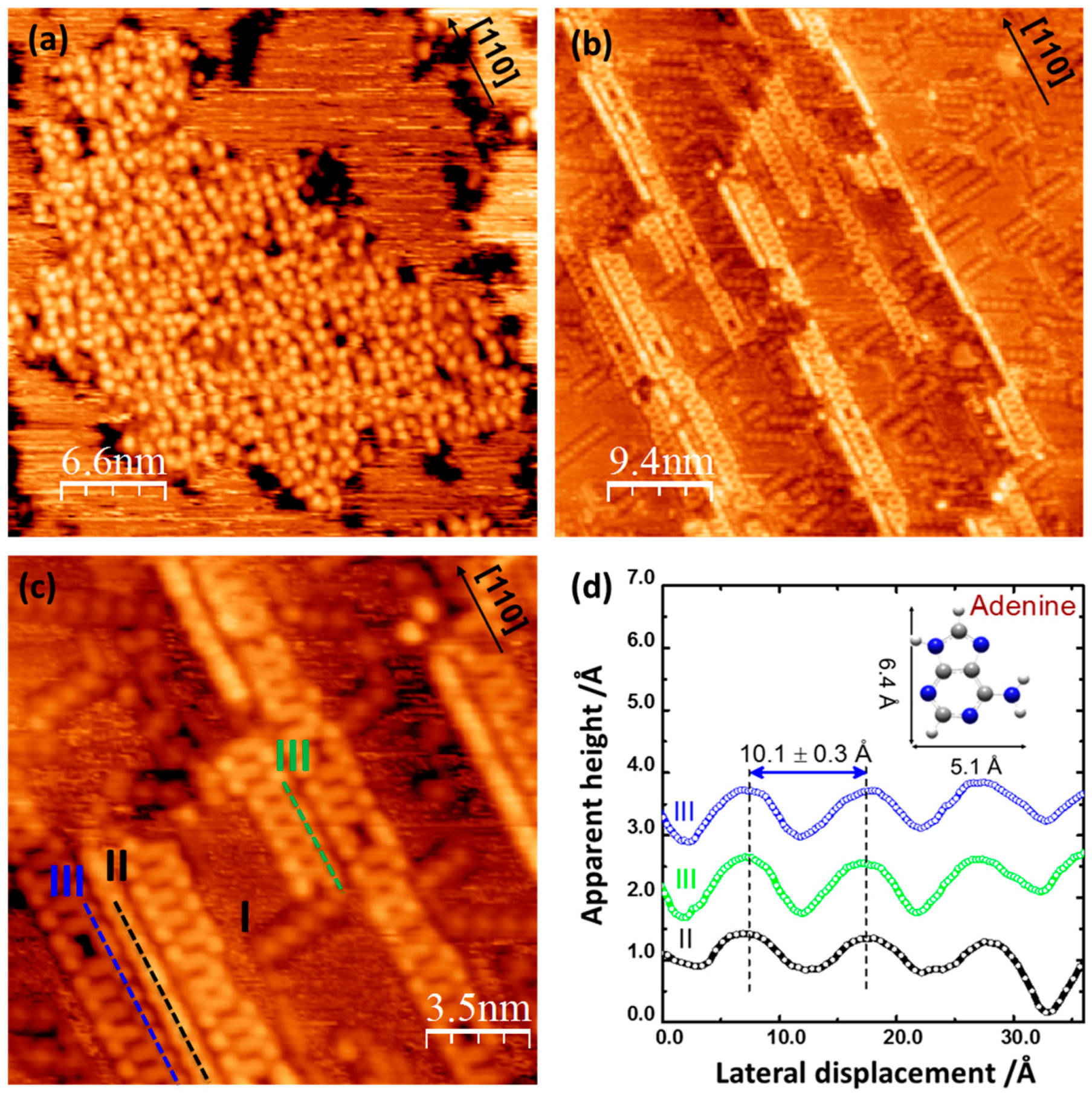
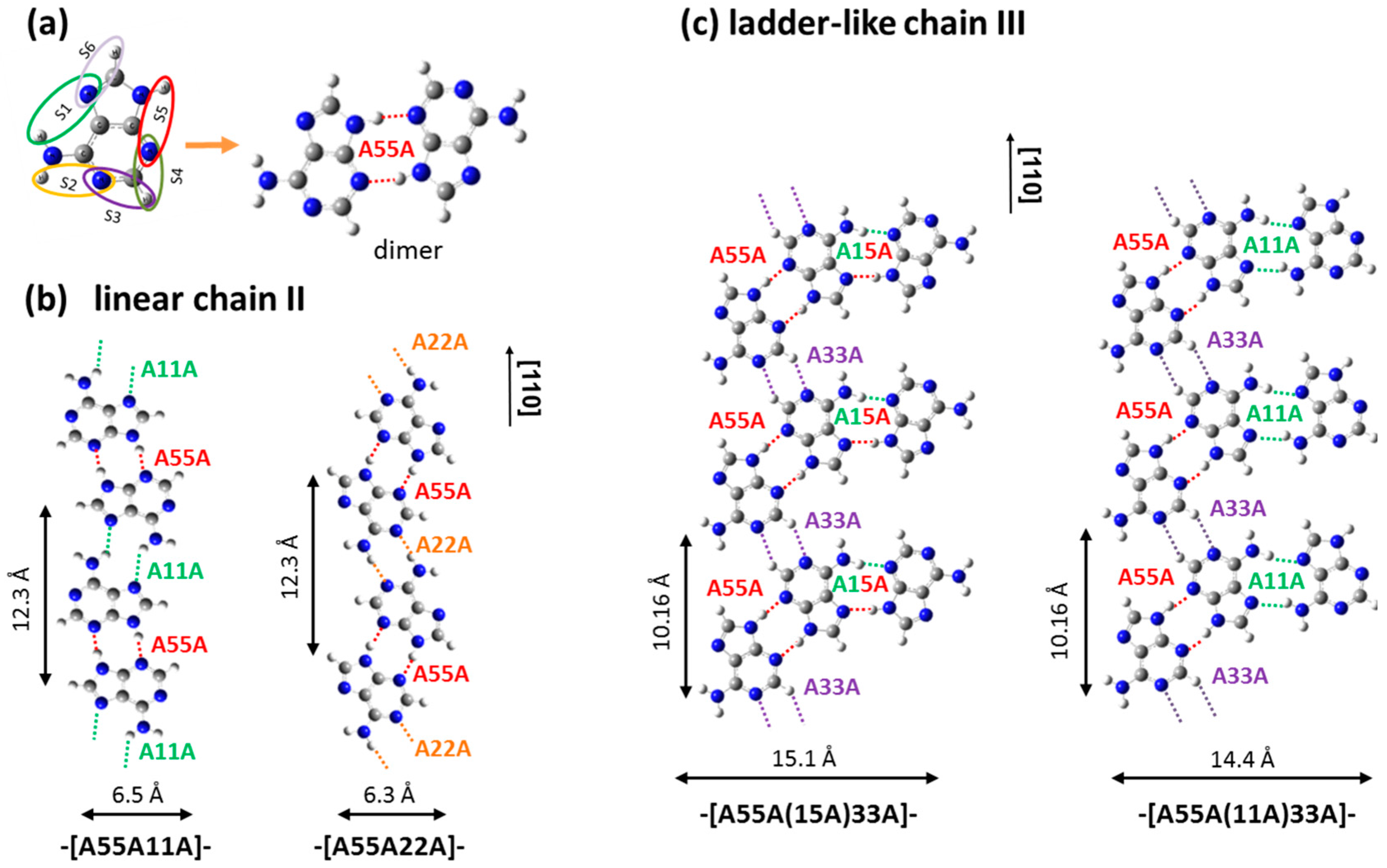
2.1. Adenine Self-Organization at Low Deposition Rate
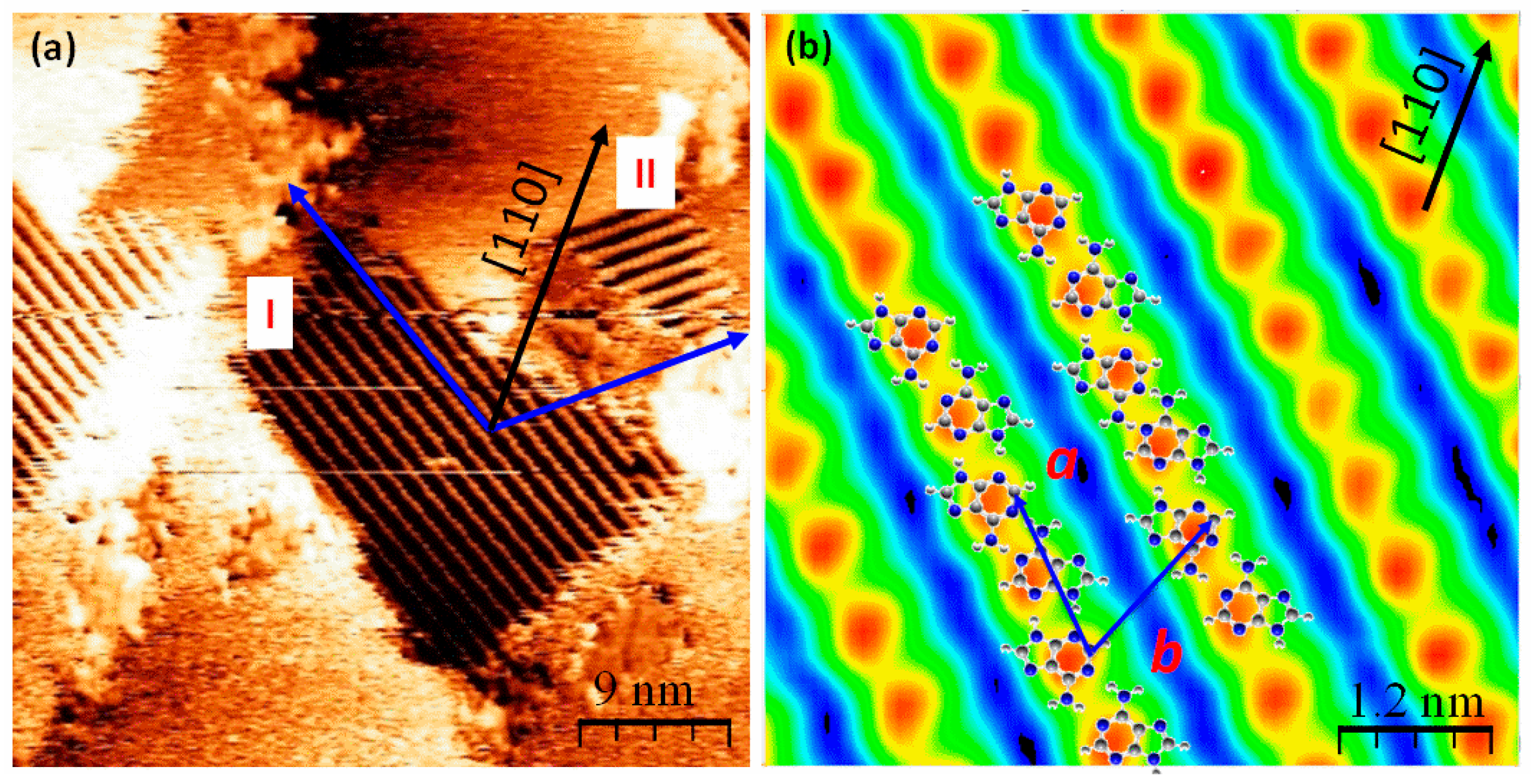
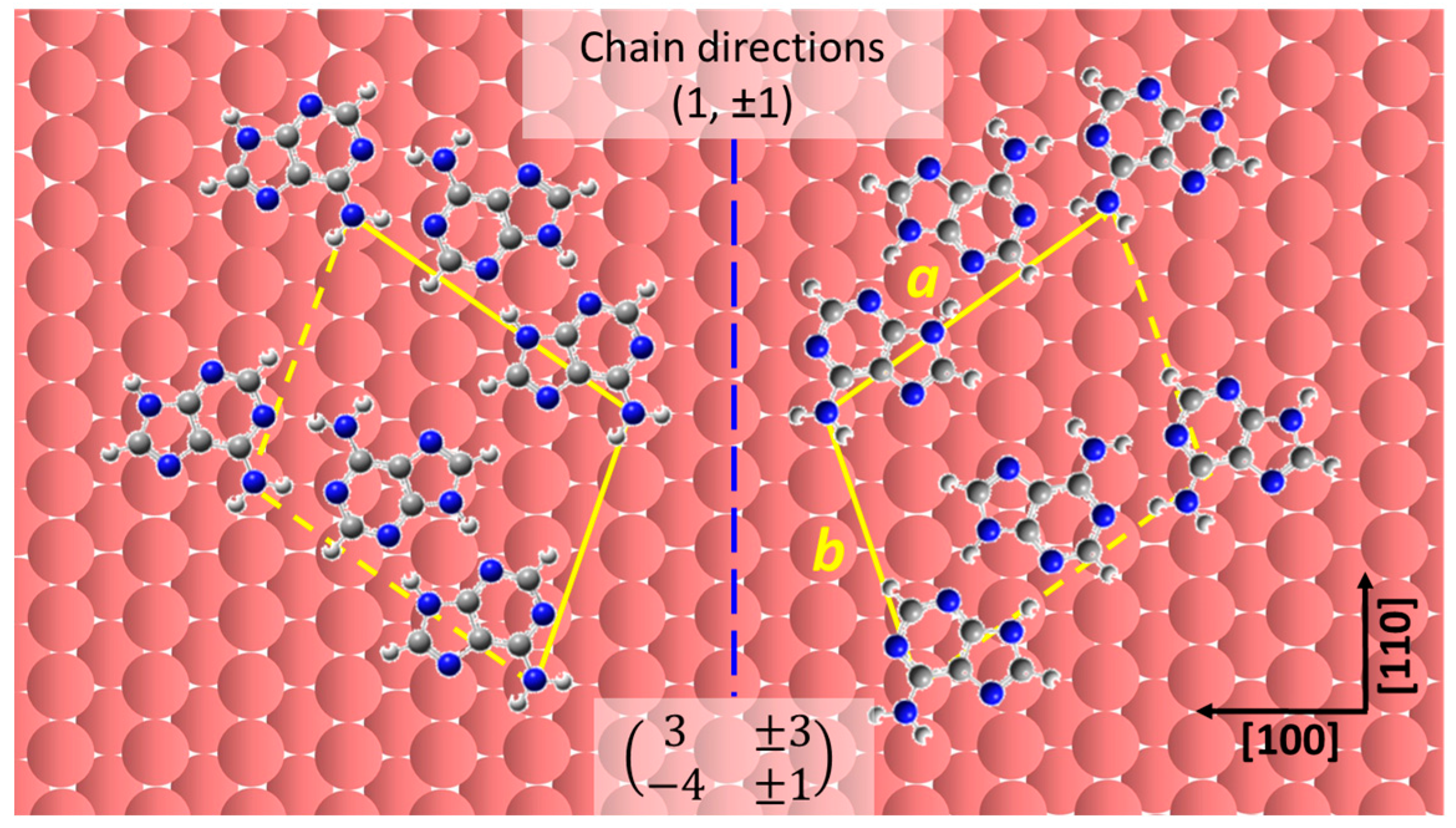
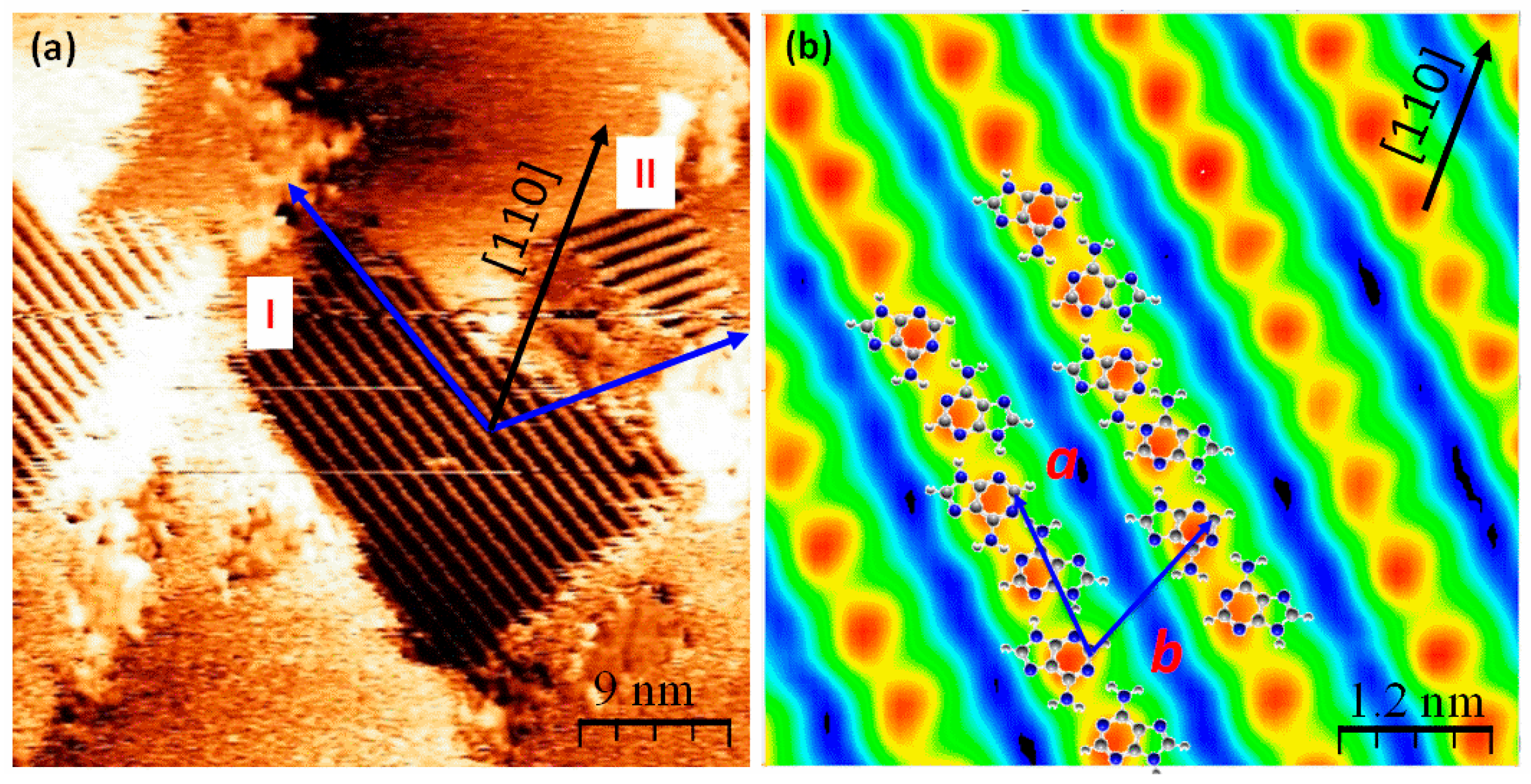
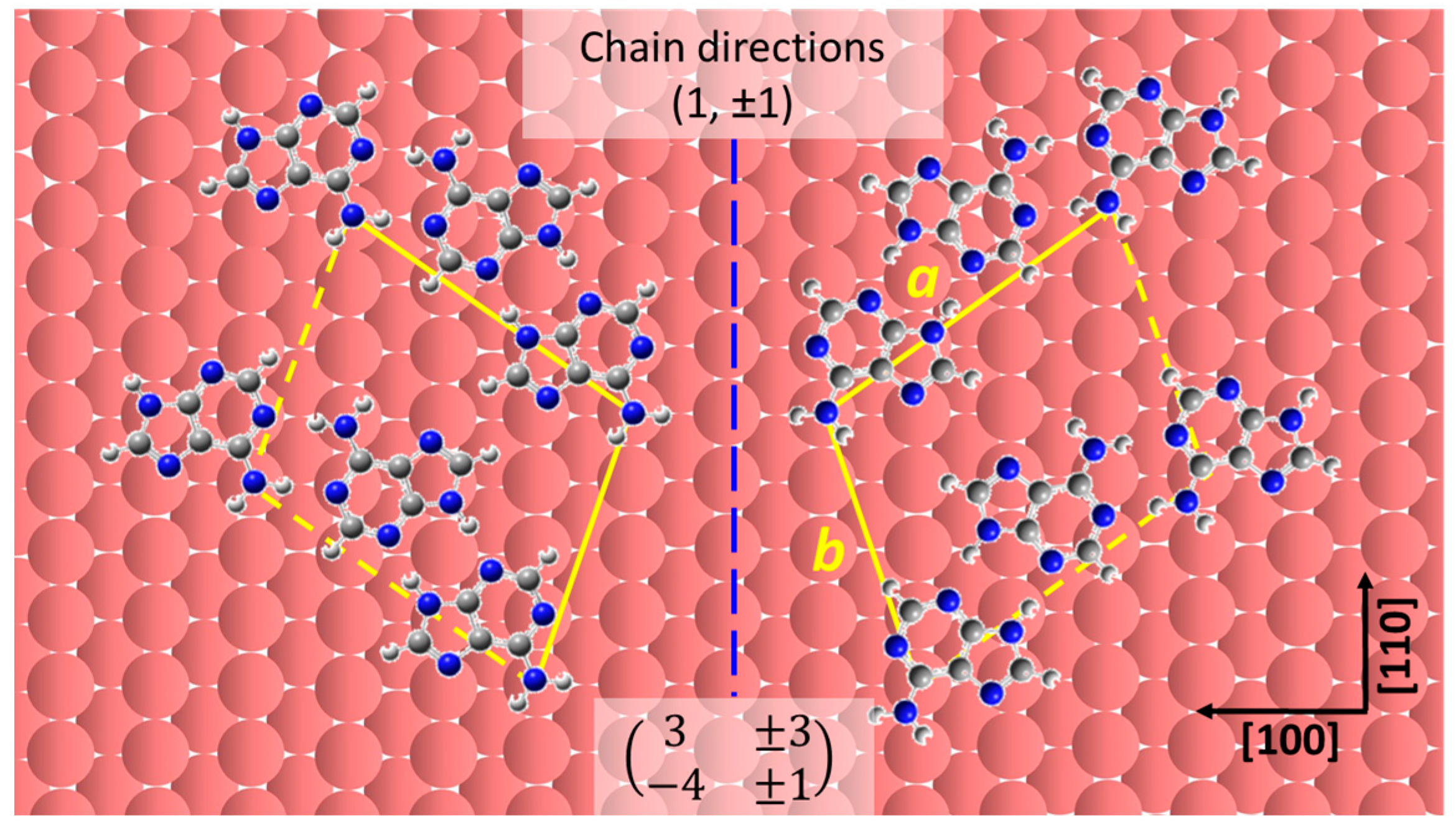
2.2. Chiral Domains Formed on a Substrate Held at 490 K
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barth, J.V.; Costantini, G.; Kern, K. Engineering atomic and molecular nanostructures at surfaces. Nature 2005, 437, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Barth, J.V. Molecular architectonic on metal surfaces. Annu. Rev. Phys. Chem. 2007, 58, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bonnesen, P.V.; Rangel, E.; Vallejo, E.; Sanchez-Castillo, A.; James Cleaves Ii, H.; Baddorf, A.P.; Sumpter, B.G.; Pan, M.; Maksymovych, P.; et al. Supramolecular polymerization of a prebiotic nucleoside provides insights into the creation of sequence-controlled polymers. Sci. Rep. 2016, 6, 18891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Wang, Y.-L.; Meng, L.; Zhang, S.B.; Gao, H.-J. Thermally controlled adenine dimer chain rotation on Cu(110): The critical role of van der waals interactions. J. Phys. Chem. C 2014, 118, 6278–6282. [Google Scholar] [CrossRef]
- Chen, Q.; Frankel, D.J.; Richardson, N.V. Self-assembly of adenine on Cu(110) surfaces. Langmuir 2002, 18, 3219–3225. [Google Scholar] [CrossRef]
- Grillo, F.; Mugnaini, V.; Oliveros, M.; Francis, S.M.; Choi, D.-J.; Rastei, M.V.; Limot, L.; Cepek, C.; Pedio, M.; Bromley, S.T.; et al. Chiral conformation at a molecular level of a propeller-like open-shell molecule on Au(111). J. Phys. Chem. Lett. 2012, 3, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Grillo, F.; Fruchtl, H.; Francis, S.M.; Mugnaini, V.; Oliveros, M.; Veciana, J.; Richardson, N.V. An ordered organic radical adsorbed on a Cu-doped Au(111) surface. Nanoscale 2012, 4, 6718–6721. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yun, K.; Lucero, A.; Huang, J.; Meng, X.; Lian, G.; Nam, H.-S.; Wallace, R.M.; Kim, M.; Venugopal, A.; et al. Low temperature synthesis of graphite on Ni films using inductively coupled plasma enhanced CVD. J. Phys. Chem. C 2015, 3, 5192–5198. [Google Scholar] [CrossRef]
- Cheng, L.; Jandhyala, S.; Mordi, G.; Lucero, A.T.; Huang, J.; Azcatl, A.; Addou, R.; Wallace, R.M.; Colombo, L.; Kim, J. Partially fluorinated graphene: Structural and electrical characterization. ACS Appl. Mater. Interfaces 2016, 8, 5002–5008. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Yamada, T.; Katano, S.; Kawai, M.; Ogasawara, H.; Nilsson, A. Geometrical characterization of adenine and guanine on Cu(110) by NEXAFS, XPS, and DFT calculation. Surf. Sci. 2007, 601, 5433–5440. [Google Scholar] [CrossRef]
- Rauls, E.; Blankenburg, S.; Schmidt, W.G. DFT calculations of adenine adsorption on coin metal (110) surfaces. Surf. Sci. 2008, 602, 2170–2174. [Google Scholar] [CrossRef]
- Kestell, J.; Boscoboinik, J.A.; Cheng, L.; Garvey, M.; Bennett, D.W.; Tysoe, W.T. Structural changes in self-catalyzed adsorption of carbon monoxide on 1,4-phenylene diisocyanide modified Au(111). J. Phys. Chem. C 2015, 119, 18317–18325. [Google Scholar] [CrossRef]
- Nelson, B.P.; Grimsrud, T.E.; Liles, M.R.; Goodman, R.M.; Corn, R.M. Surface plasmon resonance imaging measurements of DNA and RNA hybridization adsorption onto dna microarrays. Anal. Chem. 2000, 73, 1–7. [Google Scholar] [CrossRef]
- Sowerby, S.J.; Edelwirth, M.; Heckl, W.M. Self-assembly at the prebiotic solid−liquid interface: Structures of self-assembled monolayers of adenine and guanine bases formed on inorganic surfaces. J. Phys. Chem. B 1998, 102, 5914–5922. [Google Scholar] [CrossRef]
- Freund, J.E.; Edelwirth, M.; Kröbel, P.; Heckl, W.M. Structure determination of two-dimensional adenine crystals on graphite. Phys. Rev. B 1997, 55, 5394–5397. [Google Scholar] [CrossRef]
- Mamdouh, W.; Dong, M.; Kelly, R.E.A.; Kantorovich, L.N.; Besenbacher, F. Coexistence of homochiral and heterochiral adenine domains at the liquid/solid interface. J. Phys. Chem. B 2007, 111, 12048–12052. [Google Scholar] [CrossRef] [PubMed]
- Edelwirth, M.; Freund, J.; Sowerby, S.J.; Heckl, W.M. Molecular mechanics study of hydrogen bonded self-assembled adenine monolayers on graphite. Surf. Sci. 1998, 417, 201–209. [Google Scholar] [CrossRef]
- Perdigão, L.M.A.; Staniec, P.A.; Champness, N.R.; Kelly, R.E.A.; Kantorovich, L.N.; Beton, P.H. Experimental and theoretical identification of adenine monolayers on ag-terminated Si(111). Phys. Rev. B 2006, 73, 195423. [Google Scholar] [CrossRef]
- Tanaka, H.; Nakagawa, T.; Kawai, T. Two-dimensional self-assembly of dna base molecules on Cu(111) surfaces. Surf. Sci. 1996, 364, L575–L579. [Google Scholar] [CrossRef]
- Kawai, T.; Tanaka, H.; Nakagawa, T. Low dimensional self-organization of dna-base molecules on Cu(111) surfaces. Surf. Sci. 1997, 386, 124–136. [Google Scholar] [CrossRef]
- Kelly, R.E.A.; Xu, W.; Lukas, M.; Otero, R.; Mura, M.; Lee, Y.-J.; Lægsgaard, E.; Stensgaard, I.; Kantorovich, L.N.; Besenbacher, F. An investigation into the interactions between self-assembled adenine molecules and a Au(111) surface. Small 2008, 4, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Lukas, M.; Kelly, R.E.A.; Kantorovich, L.N.; Otero, R.; Xu, W.; Laegsgaard, E.; Stensgaard, I.; Besenbacher, F. Adenine monolayers on the Au(111) surface: Structure identification by scanning tunneling microscopy experiment and ab initio calculations. J. Chem. Phys. 2009, 130, 024705. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.E.A.; Kantorovich, L.N. Hexagonal adenine networks constructed from their homopairings. Surf. Sci. 2005, 589, 139–152. [Google Scholar] [CrossRef]
- McNutt, A.; Haq, S.; Raval, R. Rairs investigations on the orientation and intermolecular interactions of adenine on Cu(110). Surf. Sci. 2003, 531, 131–144. [Google Scholar] [CrossRef]
- Preuss, M.; Schmidt, W.G.; Bechstedt, F. Coulombic amino group-metal bonding: Adsorption of adenine on Cu(110). Phys. Rev. Lett. 2005, 94, 236102. [Google Scholar] [CrossRef] [PubMed]
- Preuss, M.; Bechstedt, F. Self-assembly of adenine-dimer chains on Cu(110): Driving forces from first-principles calculations. Surf. Sci. 2008, 602, 1643–1649. [Google Scholar] [CrossRef]
- Yamada, T.; Shirasaka, K.; Takano, A.; Kawai, M. Adsorption of cytosine, thymine, guanine and adenine on Cu(110) studied by infrared reflection absorption spectroscopy. Surf. Sci. 2004, 561, 233–247. [Google Scholar] [CrossRef]
- Feyer, V.; Plekan, O.; Prince, K.C.; Šutara, F.; Skála, T.; Cháb, V.; Matolín, V.; Stenuit, G.; Umari, P. Bonding at the organic/metal interface: Adenine to Cu(110). Phys. Rev. B 2009, 79, 155432. [Google Scholar] [CrossRef]
- Bussolotti, F.; Friedlein, R. Hybrid interfaces of biological molecules and metals: The prototypical case of adenine on Cu(110). J. Chem. Phys. 2010, 132, 184705. [Google Scholar] [CrossRef]
- Furukawa, M.; Tanaka, H.; Kawai, T. Formation mechanism of low-dimensional superstructure of adenine molecules and its control by chemical modification: A low-temperature scanning tunneling microscopy study. Surf. Sci. 2000, 445, 1–10. [Google Scholar] [CrossRef]
- Camargo, P.M.A.; Baumgartel, H.; Donner, C. Adsorption behaviour of DNA bases at the Au(111) electrode. PhysChemComm 2002, 5, 151–157. [Google Scholar] [CrossRef]
- Temprano, I.; Thomas, G.; Haq, S.; Dyer, M.S.; Latter, E.G.; Darling, G.R.; Uvdal, P.; Raval, R. 1D self-assembly of chemisorbed thymine on Cu(110) driven by dispersion forces. J. Chem. Phys. 2015, 142, 101916. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.E.A.; Lee, Y.J.; Kantorovich, L.N. Homopairing possibilities of the dna base adenine. J. Phys. Chem. B 2005, 109, 11933–11939. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Richardson, N.V. Enantiomeric interactions between nucleic acid bases and amino acids on solid surfaces. Nat. Mater. 2003, 2, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Horcas, I.; Fernández, R.; Gómez-Rodríguez, J.M.; Colchero, J.; Gómez-Herrero, J.; Baro, A.M. Wsxm: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03, Revision E. 01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, L. Role of Hydrogen Bonding in the Formation of Adenine Chains on Cu(110) Surfaces. Materials 2016, 9, 1016. https://doi.org/10.3390/ma9121016
Cheng L. Role of Hydrogen Bonding in the Formation of Adenine Chains on Cu(110) Surfaces. Materials. 2016; 9(12):1016. https://doi.org/10.3390/ma9121016
Chicago/Turabian StyleCheng, Lanxia. 2016. "Role of Hydrogen Bonding in the Formation of Adenine Chains on Cu(110) Surfaces" Materials 9, no. 12: 1016. https://doi.org/10.3390/ma9121016
APA StyleCheng, L. (2016). Role of Hydrogen Bonding in the Formation of Adenine Chains on Cu(110) Surfaces. Materials, 9(12), 1016. https://doi.org/10.3390/ma9121016