

Influence of a Siloxane-Modified DOPO Derivative on the Properties of Polyurethane Cationomer Coatings


Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis of TMDS–AGE
2.2. Synthesis of TMDS–AGE–DOPA
2.3. Synthesis of Polyurethane Cationomers Modified with TMDS–AGE–DOPA
3. Results and Discussion
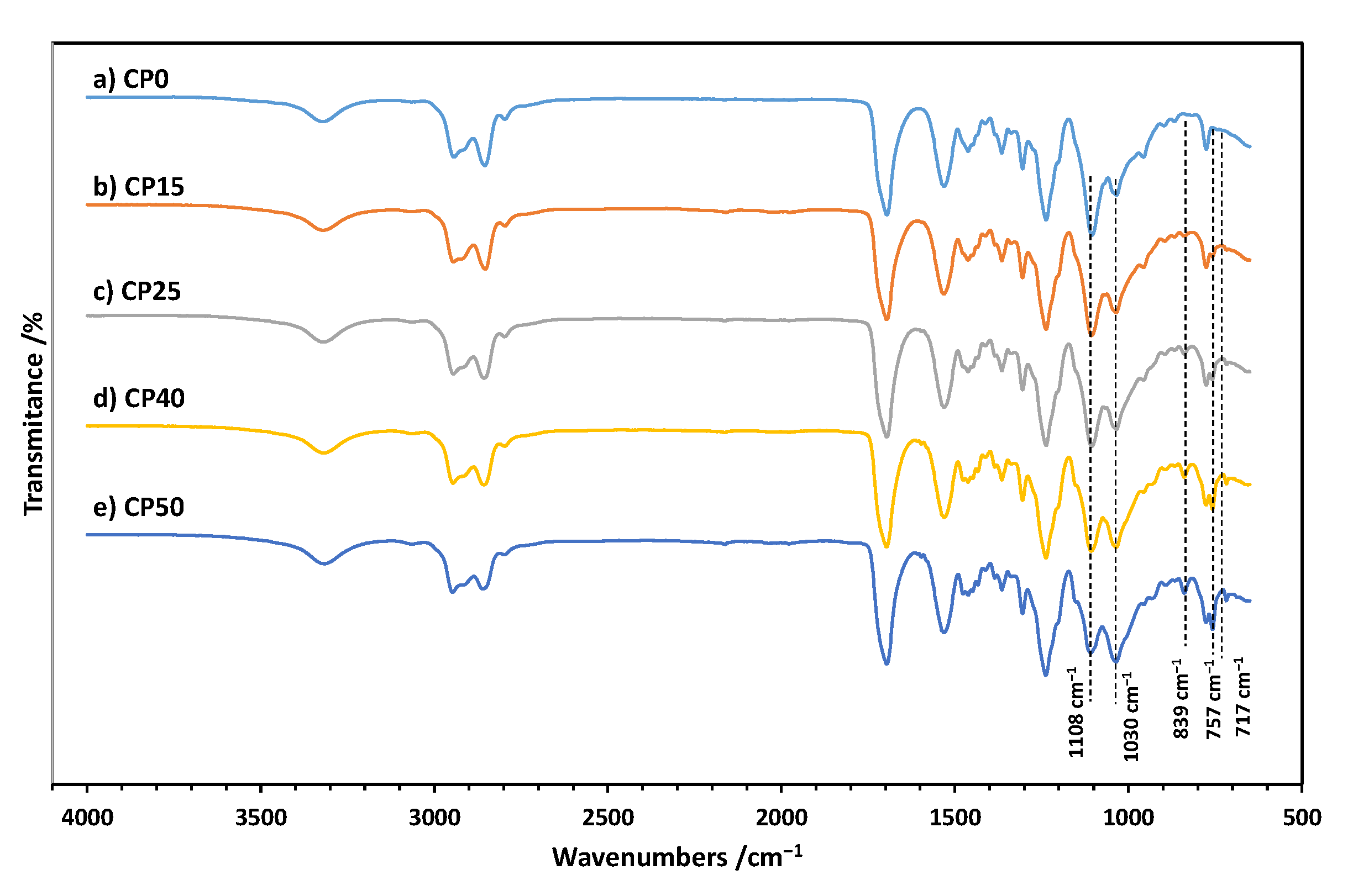
3.1. FTIR Spectra of Cationomers
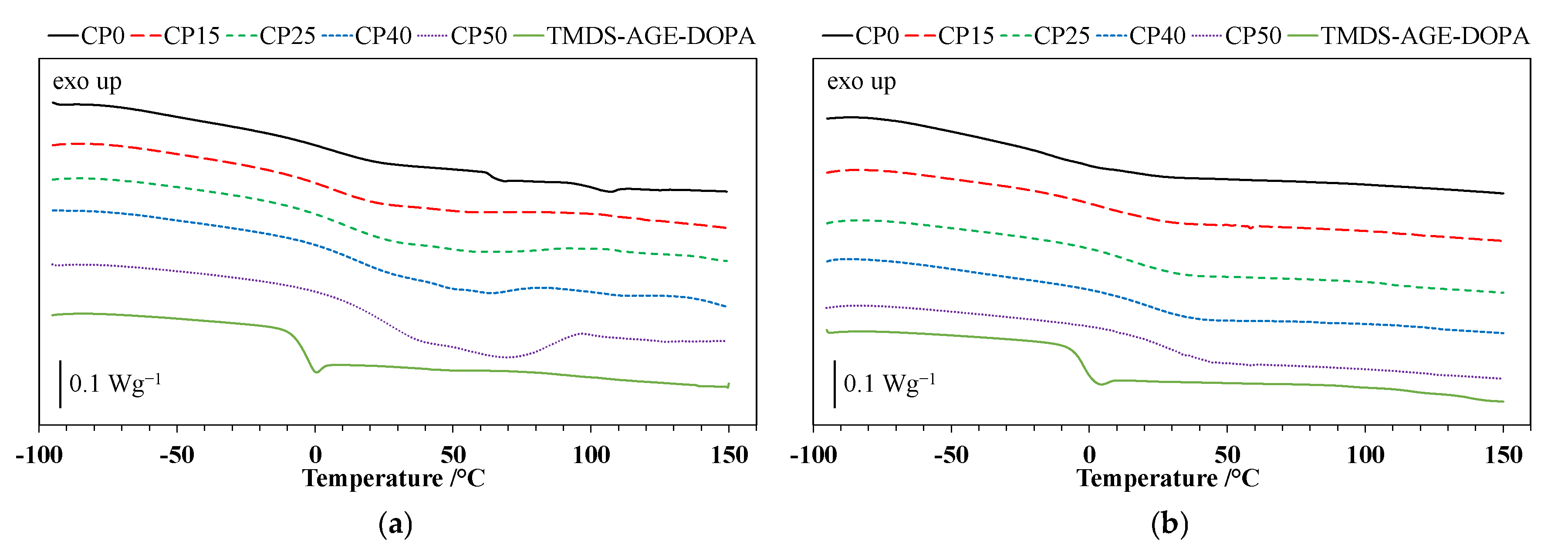
3.2. Differential Scanning Calorimetry (DSC)
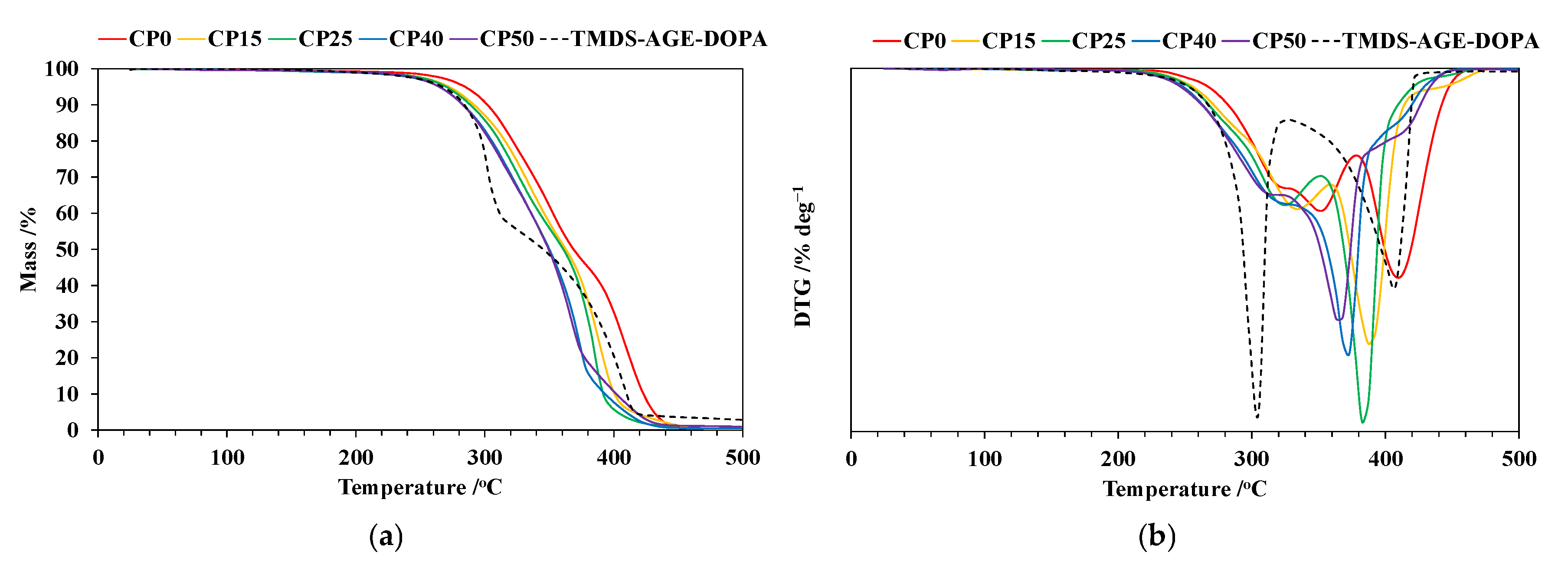
3.3. Thermogravimetric Analysis (TG)
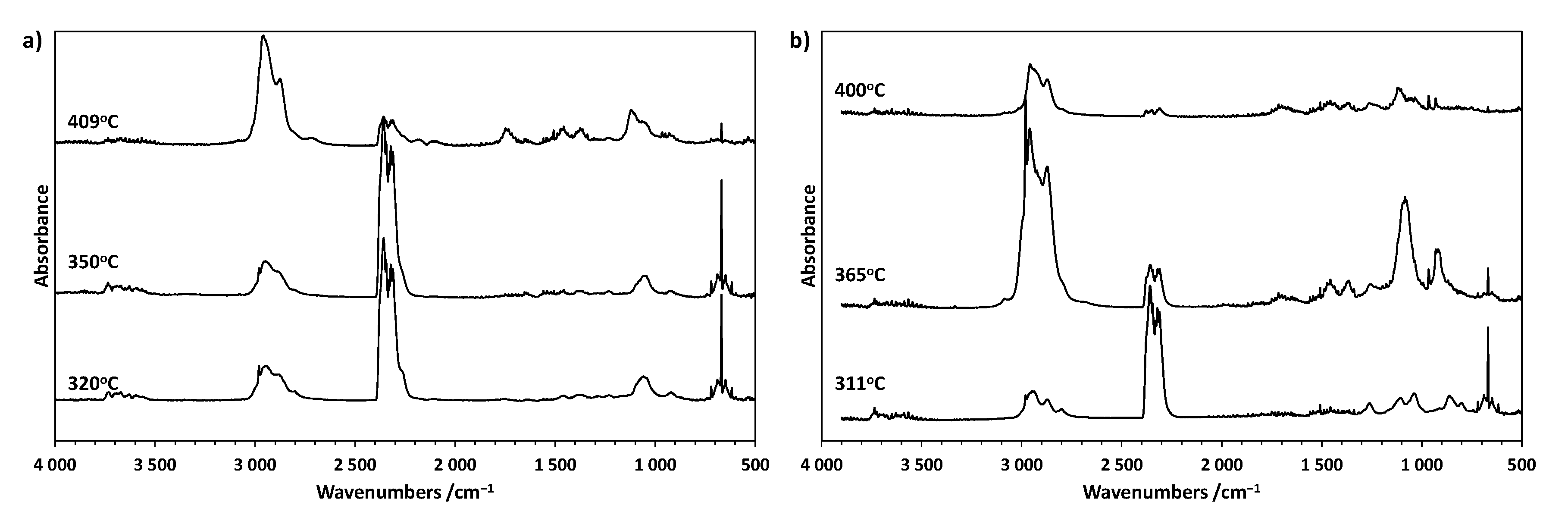
3.4. TG-FTIR Analyses
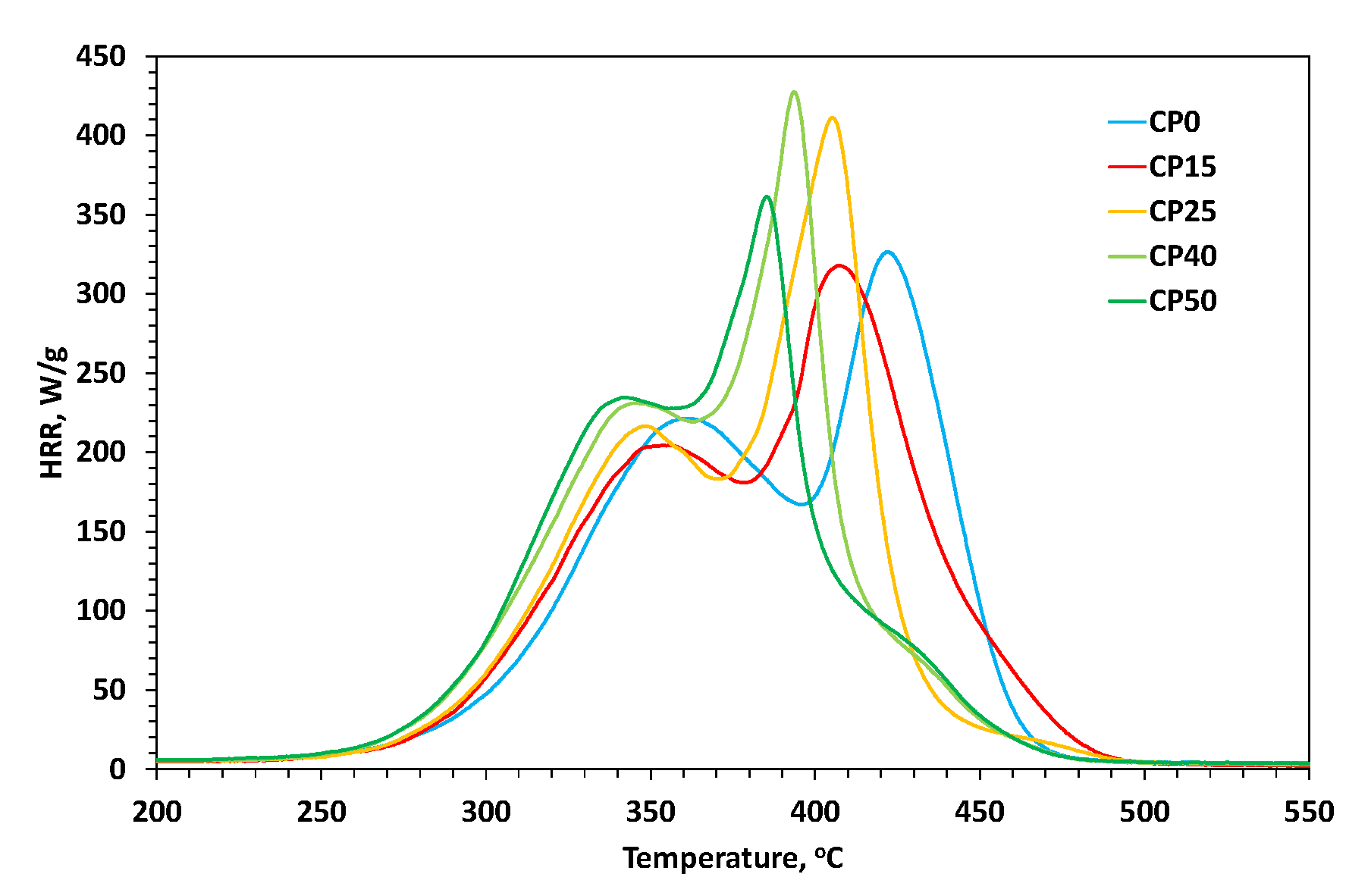
3.5. Pyrolysis-Combustion Flow Calorimetry (PCFC)
3.6. Contact Angle and Surface Free Energy
3.7. Performance Properties of the Polyurethane Cationomer Coatings
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agnol, L.D.; Dias, F.T.G.; Ornaghi, H.L., Jr.; Sangermano, M.; Bianchi, O. UV-curable waterborne polyurethane coatings: A state-of-the-art and recent advances review. Prog. Org. Coat. 2021, 154, 106156. [Google Scholar] [CrossRef]
- Morgan, A.B.; Gilman, J.W. An overview of flame retardancy of polymeric materials: Application, technology, and future directions. Fire Mater. 2013, 37, 259–279. [Google Scholar] [CrossRef]
- Wen, O.Y.; Tohir, M.Z.M.; Yeaw, T.C.S.; Razak, M.A.; Zainuddin, H.S.; Hamid, M.R.A. Fire-resistant and flame-retardant surface finishing of polymers and textiles: A state-of-the-art review. Prog. Org. Coat. 2023, 175, 107330. [Google Scholar] [CrossRef]
- Laoutid, F.; Bonnaud, L.; Alexandre, M.; Lopez-Cuesta, J.M.; Dubois, P. New prospects in flame retardant polymer materials: From fundamentals to nanocomposites. Mater. Sci. Eng. R 2009, 63, 100–125. [Google Scholar] [CrossRef]
- Lu, S.-Y.; Hamerton, I. Recent developments in the chemistry of halogen-free flame retardant polymers. Prog. Polym. Sci. 2002, 27, 1661–1712. [Google Scholar] [CrossRef]
- Varganici, C.D.; Rosu, L.; Bifulco, A.; Rosu, D.; Mustata, F.; Gaan, S. Recent advances in flame retardant epoxy systems from reactive DOPO–based phosphorus additives. Polym. Degrad. Stab. 2022, 202, 110020. [Google Scholar] [CrossRef]
- Feng, L.; Wang, W.; Song, B.; Zhu, X.; Wang, L.; Shao, R.; Li, T.; Pei, X.; Wang, L.; Qian, X.; et al. Synthesis of P, N and Si-containing waterborne polyurethane with excellent flame retardant, alkali resistance and flexibility via one-step synthetic approach. Prog. Org. Coat. 2023, 174, 107286. [Google Scholar] [CrossRef]
- Parcheta-Szwindowska, P.; Habaj, J.; Krzemińska, I.; Datta, J. A Comprehensive Review of Reactive Flame Retardants for Polyurethane Materials: Current Development and Future Opportunities in an Environmentally Friendly Direction. Int. J. Mol. Sci. 2024, 25, 5512. [Google Scholar] [CrossRef]
- Yin, X.; Li, L.; Pang, H.; Luo, Y.; Zhang, B. Halogen-free instinct flame-retardant waterborne polyurethanes: Composition, performance, and application. RSC Adv. 2022, 12, 14509. [Google Scholar] [CrossRef]
- Ma, K.; Chen, Q.; Li, Y.; You, Y.; Zou, G.; Xie, H. Atom-Economic Synthesis of Phosphorus-Containing Biobased Aromatic Diol for Flame-Retardant Robust Polyurethane. ACS Appl. Polym. Mater. 2024, 6, 4830–4839. [Google Scholar] [CrossRef]
- Liu, X.; Salmeia, K.A.; Rentsch, D.; Hao, J.; Gaan, S. Thermal decomposition and flammability of rigid PU foams containing some DOPO derivatives and other phosphorus compounds. J. Anal. Appl. Pyrolysis 2017, 124, 219–229. [Google Scholar] [CrossRef]
- Ding, N.; Yang, Y.; Lu, B.; Zhang, R.; Xu, P.; Niu, D.; Yang, W.; Ma, P. From a bio-based polyphenol diol intermedia to high-performance polyurethane elastomer: Thermal stability, reprocessability and flame retardancy. J. Colloid Interface Sci. 2025, 680, 608–617. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, H.; Wang, H.; Cheng, X.; Du, Z. Enhanced flame retardancy and mechanical properties of waterborne polyurethane based on the phosphorus and nitrogen containing polybutadiene acrylonitrile. J. Appl. Polym. Sci. 2021, 138, e50432. [Google Scholar] [CrossRef]
- Qu, J.; Leng, G.; Yu, H.; Li, G.; Wang, S.; Weng, Y.; Duan, B.; Liu, J. Flame-retardant waterborne polyurethane based on the synergistic effect of HGB and DOPO derivatives. J. Coat. Technol. Res. 2024, 22, 369–383. [Google Scholar] [CrossRef]
- Zhu, X.; Cheng, F.; Si, Y.; Wang, J.; Wang, P.; Shi, C.; Han, J. The impact of DOPO-derived phosphoramides containing triazine moieties on the flame retardancy and smoke suppression properties of waterborne polyurethane. J. Polym. Sci. 2024, 62, 4058–4071. [Google Scholar] [CrossRef]
- Wang, H.; Wang, S.; Du, X.; Wang, H.; Cheng, X.; Du, Z. Synthesis of a novel flame retardant based on DOPO derivatives and its application in waterborne polyurethane. RSC Adv. 2019, 9, 7411. [Google Scholar] [CrossRef]
- Zhou, J.; Dong, F.; Liu, C.; Lu, K. P, N, Si synergistic flame-retarding water polyurethane coating with superior flame retardancy and hydrophobicity. J. Polym. Res. 2023, 30, 260. [Google Scholar] [CrossRef]
- Cui, M.; Li, J.; Chen, X.; Hong, W.; Chen, Y.; Xiang, J.; Yan, J.; Fan, H. A halogen-free, flame retardant, waterborne polyurethane coating based on the synergistic effect of phosphorus and silicon. Prog. Org. Coat. 2021, 158, 106359. [Google Scholar] [CrossRef]
- Byczyński, Ł.; Szołyga, M.; Kisiel, M. Poly(urethane-acrylate) cationomer coatings with increased hydrophobicity and reduced flammability. Prog. Org. Coat. 2024, 186, 108008. [Google Scholar] [CrossRef]
- Szołyga, M.; Dutkiewicz, M.; Nowicki, M.; Sałasińska, K.; Celiński, M.; Marciniec, B. Phosphorus-containing silsesquioxane derivatives as additive or reactive components of epoxy resins. Materials 2020, 13, 5373. [Google Scholar] [CrossRef]
- van Oss, C.J.; Chaudhury, M.K.; Good, R.J. Interfacial Lifshitz-van der Waals and polar interactions in macroscopic systems. Chem. Rev. 1988, 88, 927–941. [Google Scholar] [CrossRef]
- Owens, D.K.; Wendt, R.C. Estimation of the surface free energy of polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- EN ISO 2813:2001; Paints and Varnishes—Determination of Gloss Value at 20°, 60° and 85°. ISO: Geneva, Switzerland, 2001.
- EN ISO 1522:2001; Paints and Varnishes—Pendulum Damping Test. ISO: Geneva, Switzerland, 2001.
- EN ISO 6272:2011; Paints and Varnishes—Rapid-Deformation (Impact Resistance) Tests. ISO: Geneva, Switzerland, 2011.
- Zhu, M.; Ma, Z.; Liu, L.; Zhang, J.; Huo, S.; Song, P. Recent advances in fire-retardant rigid polyurethane foam. J. Mater. Sci. Technol. 2022, 112, 315–328. [Google Scholar] [CrossRef]
- Dutkiewicz, M.; Przybylak, M.; Januszewski, R.; Maciejewski, H. Synthesis and flame retardant efficacy of hexakis(3-(triethoxysilyl)propyloxy)cyclotriphosphazene/silica coatings for cotton fabrics. Polym. Degrad. Stab. 2018, 148, 10–18. [Google Scholar] [CrossRef]

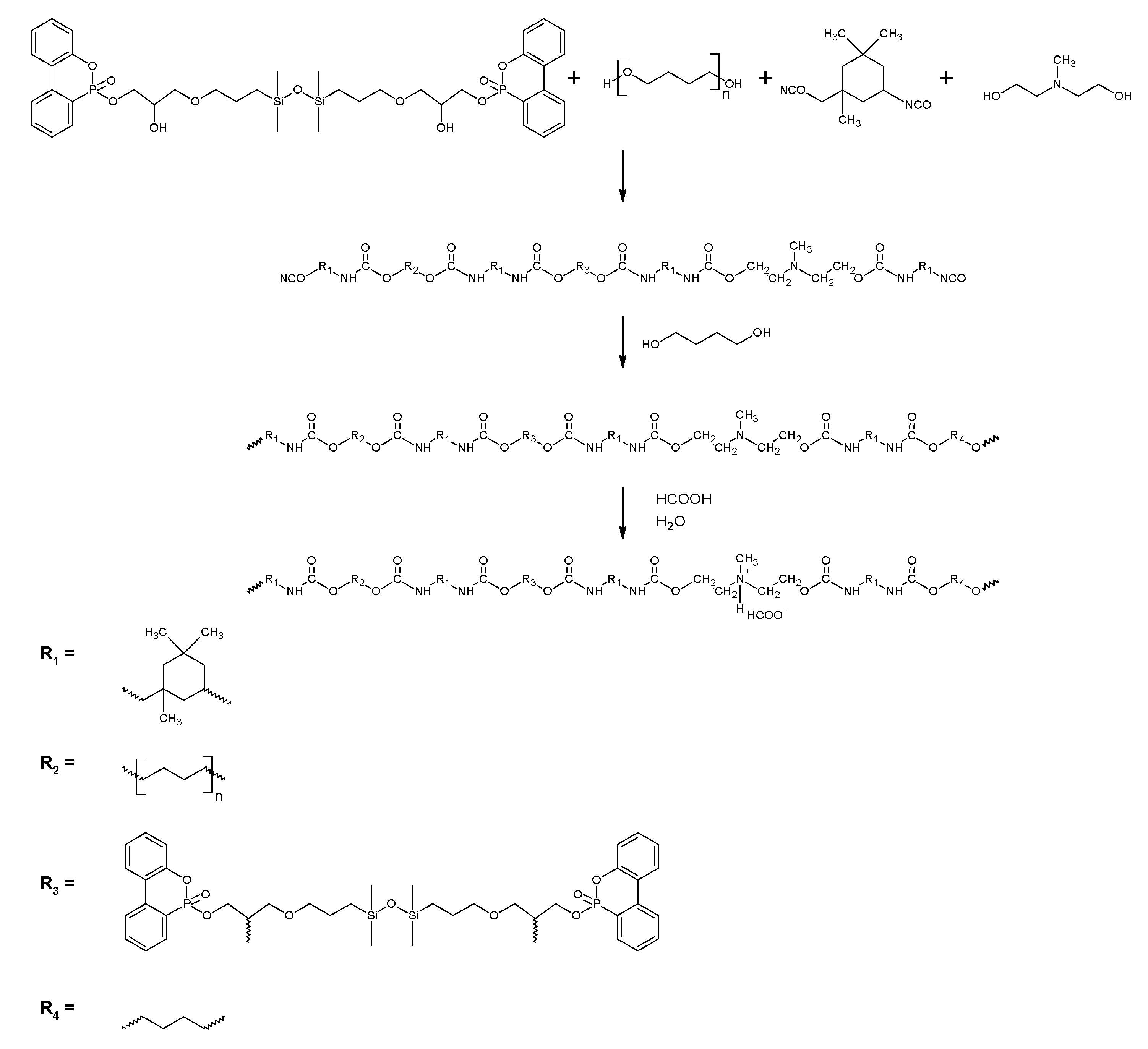


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CP0 | CP15 | CP25 | CP40 | CP50 |
|---|---|---|---|---|---|
| IPDI/mol | 4 | 4 | 4 | 4 | 4 |
| PTMO/mol | 1 | 0.85 | 0.75 | 0.60 | 0.50 |
| TMDS–AGE–DOPA/mol | 0 | 0.15 | 0.25 | 0.40 | 0.50 |
| NMDA/mol | 1 | 1 | 1 | 1 | 1 |
| HCOOH/mol | 1 | 1 | 1 | 1 | 1 |
| BD/mol | 2 | 2 | 2 | 2 | 2 |
| IPDI/wt.% | 40% | 40% | 41% | 41% | 41% |
| PTMO/wt.% | 45% | 38% | 34% | 28% | 23% |
| TMDS–AGE–DOPA/wt.% | 0% | 6% | 9% | 15% | 19% |
| NMDA/wt.% | 5% | 5% | 5% | 6% | 6% |
| HCOOH/wt.% | 2% | 2% | 2% | 2% | 2% |
| BD/wt.% | 8% | 8% | 8% | 8% | 8% |
| Sample | TMDS–AGE–DOPA | CP0 | CP15 | CP25 | CP40 | CP50 |
|---|---|---|---|---|---|---|
| Tg/°C (1st heating) | −5 | 1 | 2 | 8 | 13 | 22 |
| Tg/°C (2nd heating) | −3 | −19 | 3 | 12 | 17 | 25 |
| Tonset/°C | 271 | 288 | 276 | 272 | 268 | 264 |
| T5%/°C | 265 | 283 | 270 | 268 | 262 | 261 |
| Tmax1/°C | 301 | 351 | 333 | 323 | - | - |
| Δm1/% | 44.5 | 24.3 * | 47.7 | 44.9 | 36.1 * | 31.1 * |
| 30.4 | ||||||
| Tmax2/°C | 407 | 409 | 388 | 382 | 370 | 365 |
| Δm2/% | 53.0 | 44.8 | 48.7 3.0 * | 53.6 1.0 * | 52.2 11.1 * | 53.2 14.6 * |
| Remaining mass/% | 2.5 | 0.5 | 0.6 | 0.5 | 0.6 | 1.1 |
| Sample | Peak I pHRR [W/g] | Peak I TPHRR [°C] | Peak II pHRR [W/g] | Peak II TPHRR [°C] | Peak III pHRR [W/g] | Peak III TPHRR [°C] | ηc [J/g·K] | THR [kJ/g] |
|---|---|---|---|---|---|---|---|---|
| CP0 | 221 | 361 | 327 | 422 | - | - | 474 | 29.3 |
| CP15 | 205 | 356 | 318 | 408 | - | - | 382 | 29.5 |
| CP25 | 217 | 348 | 411 | 405 | - | - | 500 | 28.6 |
| CP40 | 231 | 346 | 427 | 394 | 74 | 428 | 475 | 28.7 |
| CP50 | 235 | 343 | 361 | 385 | 78 | 430 | 530 | 27.8 |
| Sample | CP0 | CP15 | CP25 | CP40 | CP50 |
|---|---|---|---|---|---|
| Diiodomethane CA/deg | 43 | 41 | 40 | 35 | 30 |
| Formamide CA/deg | 79 | 70 | 69 | 58 | 59 |
| Water CA/deg | 89 | 87 | 87 | 86 | 85 |
| SFE parameters—vOG method | |||||
| γSLW/mJ m−2 | 37.9 | 37.2 | 37.7 | 41.8 | 44.0 |
| γS+/mJ m−2 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 |
| γS−/mJ m−2 | 11.0 | 2.6 | 3.3 | 2.1 | 3.0 |
| γSAB/mJ m−2 | 0.0 | 0.0 | 0.0 | 0.8 | 0.0 |
| γS/mJ m−2 | 37.9 | 37.2 | 37.7 | 42.6 | 44.0 |
| SFE parameters—OW method Water-Diiodomethane liquids | |||||
| γSd/mJ m−2 | 1.2 | 0.7 | 1.0 | 0.9 | 0.9 |
| γSp/mJ m−2 | 36.4 | 38.0 | 38.0 | 40.6 | 43.1 |
| γS/mJ m−2 | 37.6 | 38.7 | 39.0 | 41.5 | 43.9 |
| SFE parameters—OW method Formamide-Diiodomethane liquids | |||||
| γSd/mJ m−2 | 0.0 | 1.5 | 1.7 | 4.8 | 3.4 |
| γSp/mJ m−2 | 39.0 | 36.8 | 37.2 | 36.9 | 40.3 |
| γS/mJ m−2 | 39.0 | 38.3 | 38.9 | 41.7 | 43.7 |
| Sample | CP0 | CP15 | CP25 | CP40 | CP50 |
|---|---|---|---|---|---|
| Gloss at 20°/GU | 138 | 173 | 161 | 153 | 136 |
| Gloss at 60°/GU | 146 | 166 | 159 | 154 | 144 |
| Gloss at 85°/GU | 101 | 110 | 104 | 107 | 102 |
| Persoz hardness/- | 0.05 | 0.07 | 0.10 | 0.19 | 0.32 |
| Impact resistance/cm | 100 | 100 | 50 | 25 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byczyński, Ł.; Szołyga, M.; Król, P. Influence of a Siloxane-Modified DOPO Derivative on the Properties of Polyurethane Cationomer Coatings. Materials 2025, 18, 789. https://doi.org/10.3390/ma18040789
Byczyński Ł, Szołyga M, Król P. Influence of a Siloxane-Modified DOPO Derivative on the Properties of Polyurethane Cationomer Coatings. Materials. 2025; 18(4):789. https://doi.org/10.3390/ma18040789
Chicago/Turabian StyleByczyński, Łukasz, Mariusz Szołyga, and Piotr Król. 2025. "Influence of a Siloxane-Modified DOPO Derivative on the Properties of Polyurethane Cationomer Coatings" Materials 18, no. 4: 789. https://doi.org/10.3390/ma18040789
APA StyleByczyński, Ł., Szołyga, M., & Król, P. (2025). Influence of a Siloxane-Modified DOPO Derivative on the Properties of Polyurethane Cationomer Coatings. Materials, 18(4), 789. https://doi.org/10.3390/ma18040789