
Silicon Oxycarbide Thin Films Produced by Hydrogen-Induced CVD Process from Cyclic Dioxa-Tetrasilacyclohexane

 , , , , and
, , , , and
Abstract
1. Introduction
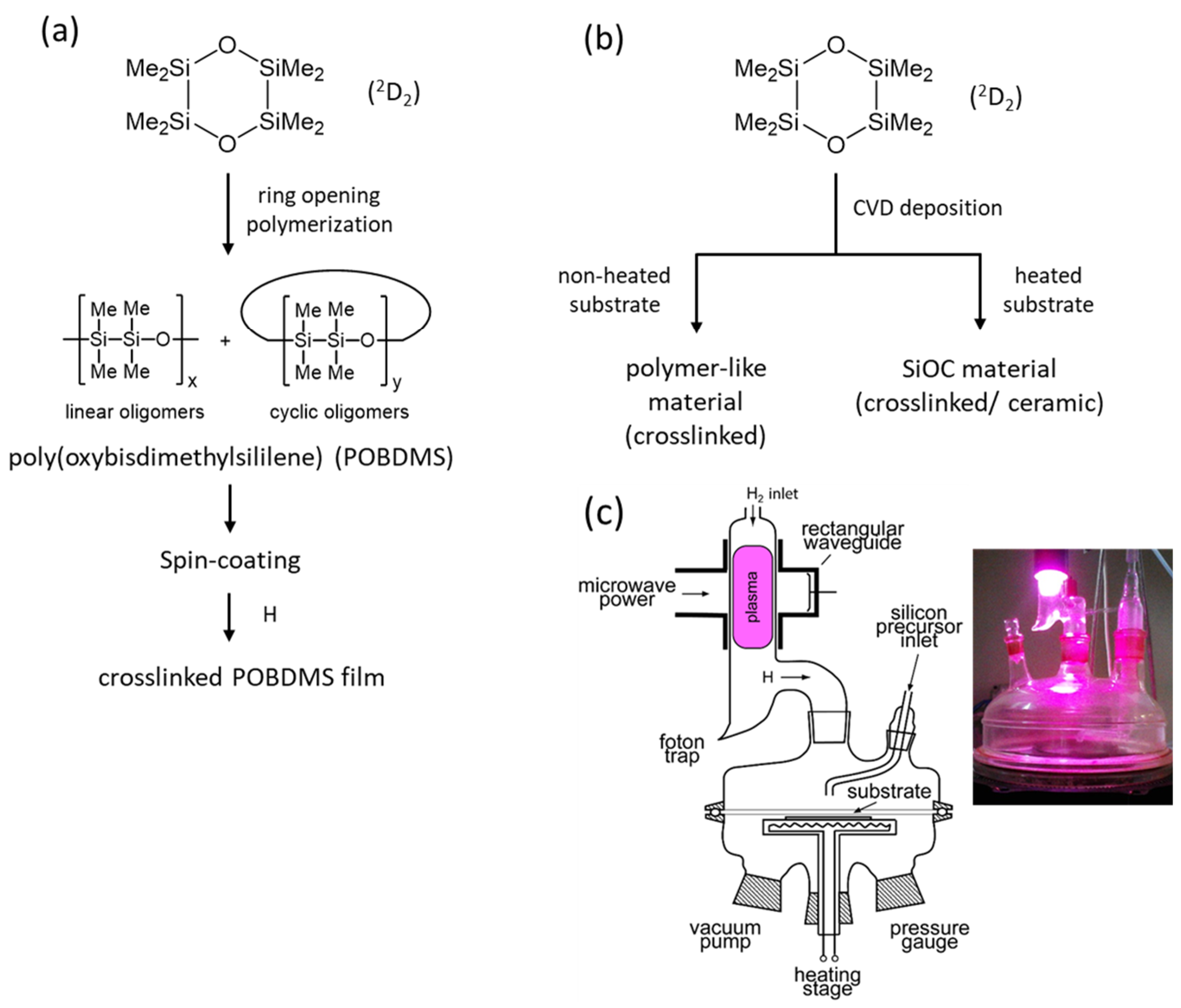
2. Materials and Methods
3. Results and Discussion
3.1. Properties of HRP-Treated POBDMS Coatings
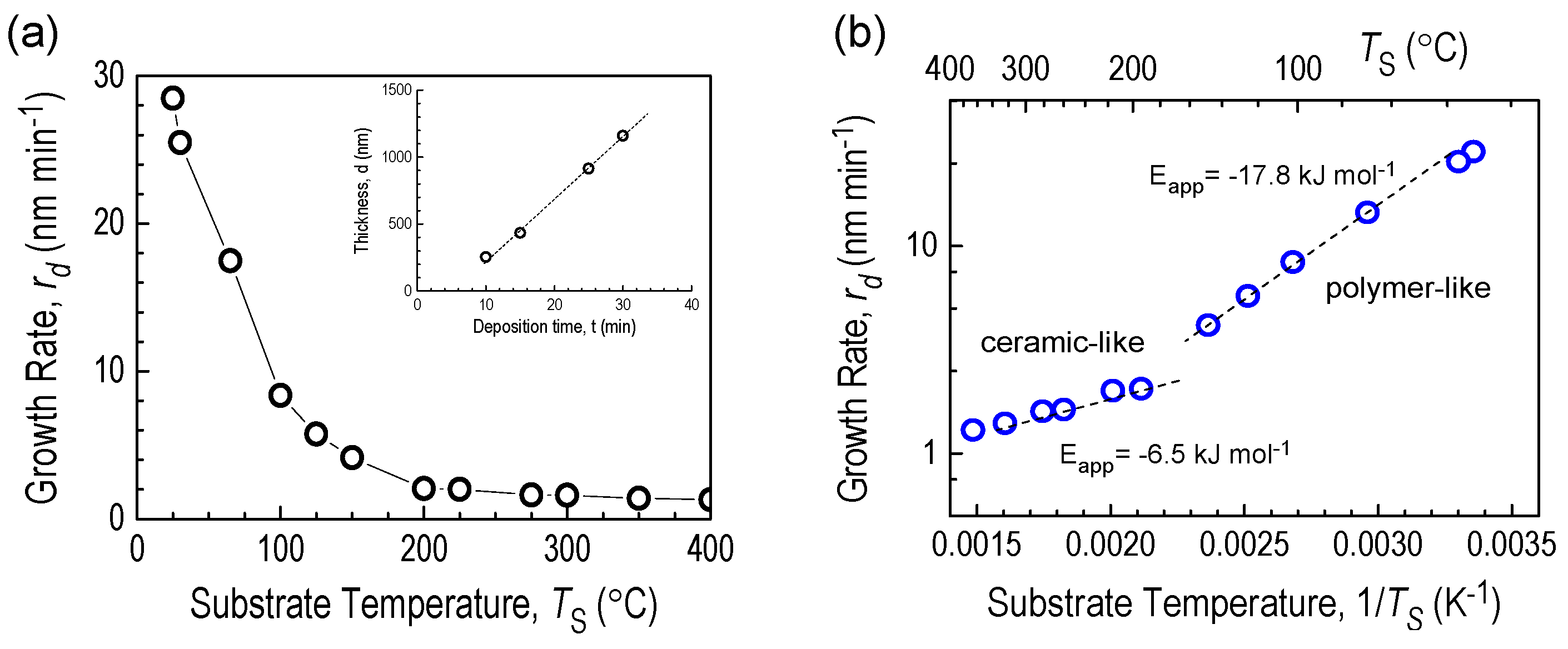
3.2. Film Deposition Rates in RHP-CVD Process from 2D2 Precursor
3.3. Chemical Structure of the CVD Films
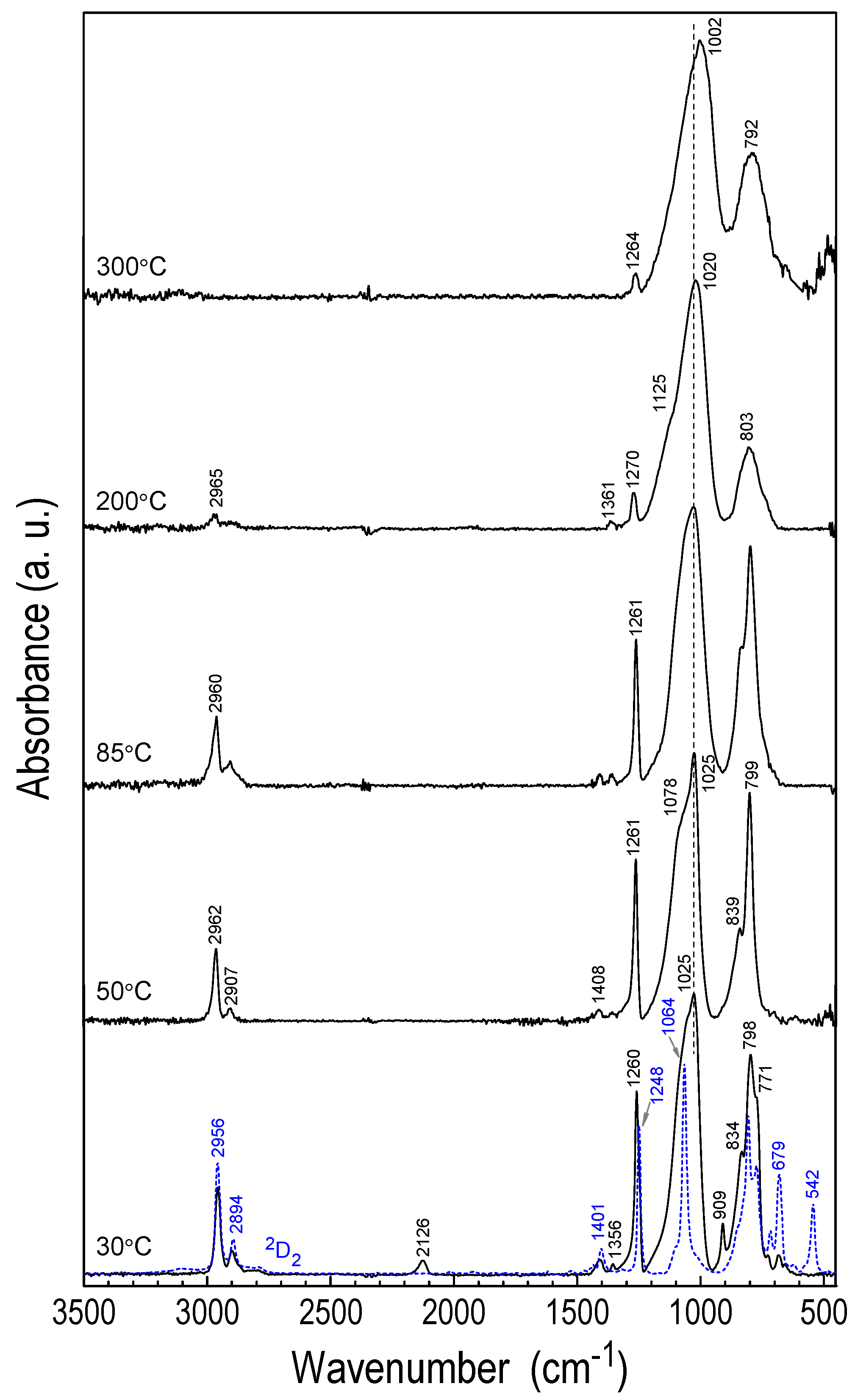
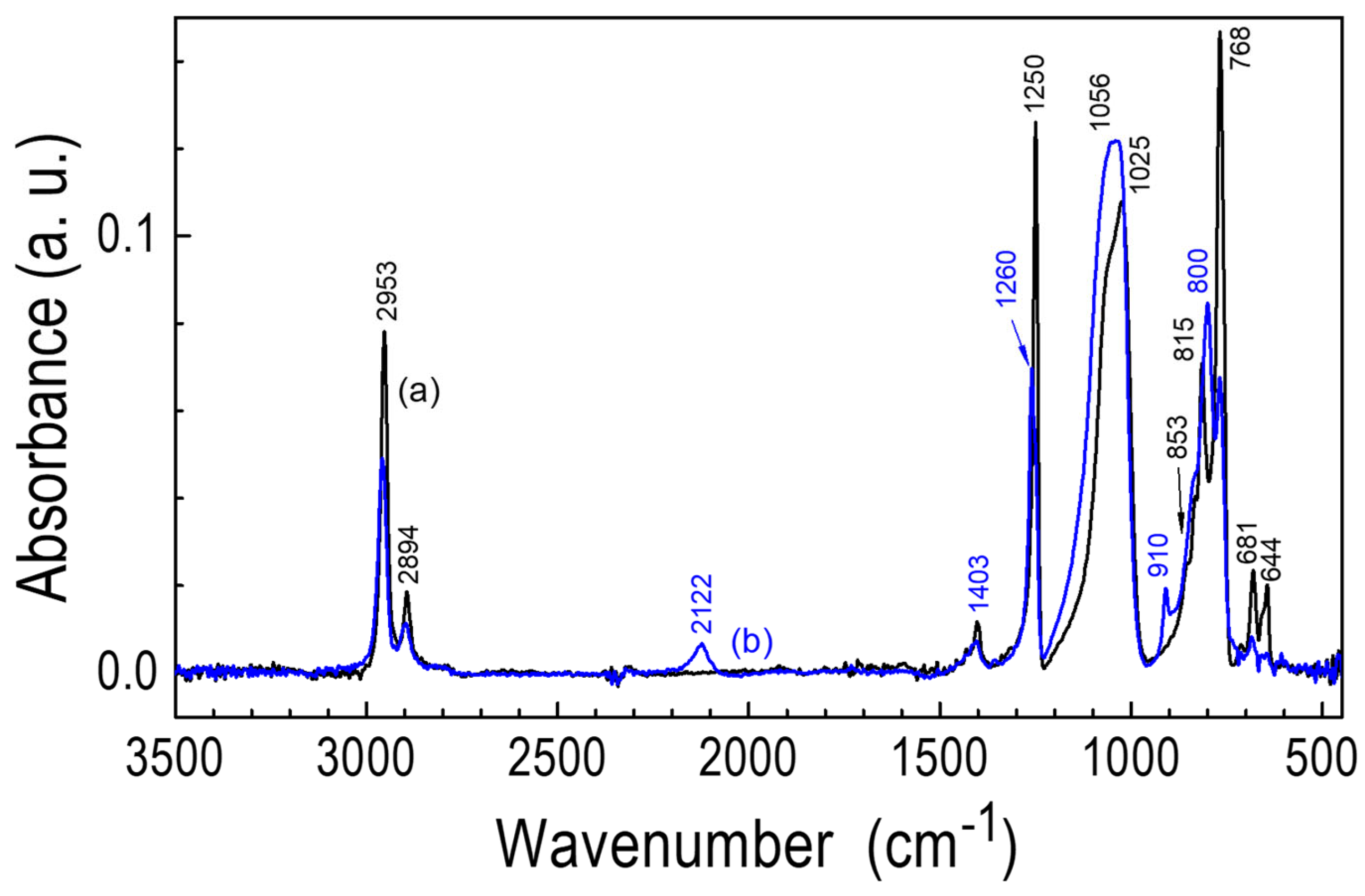
3.3.1. FTIR Studies
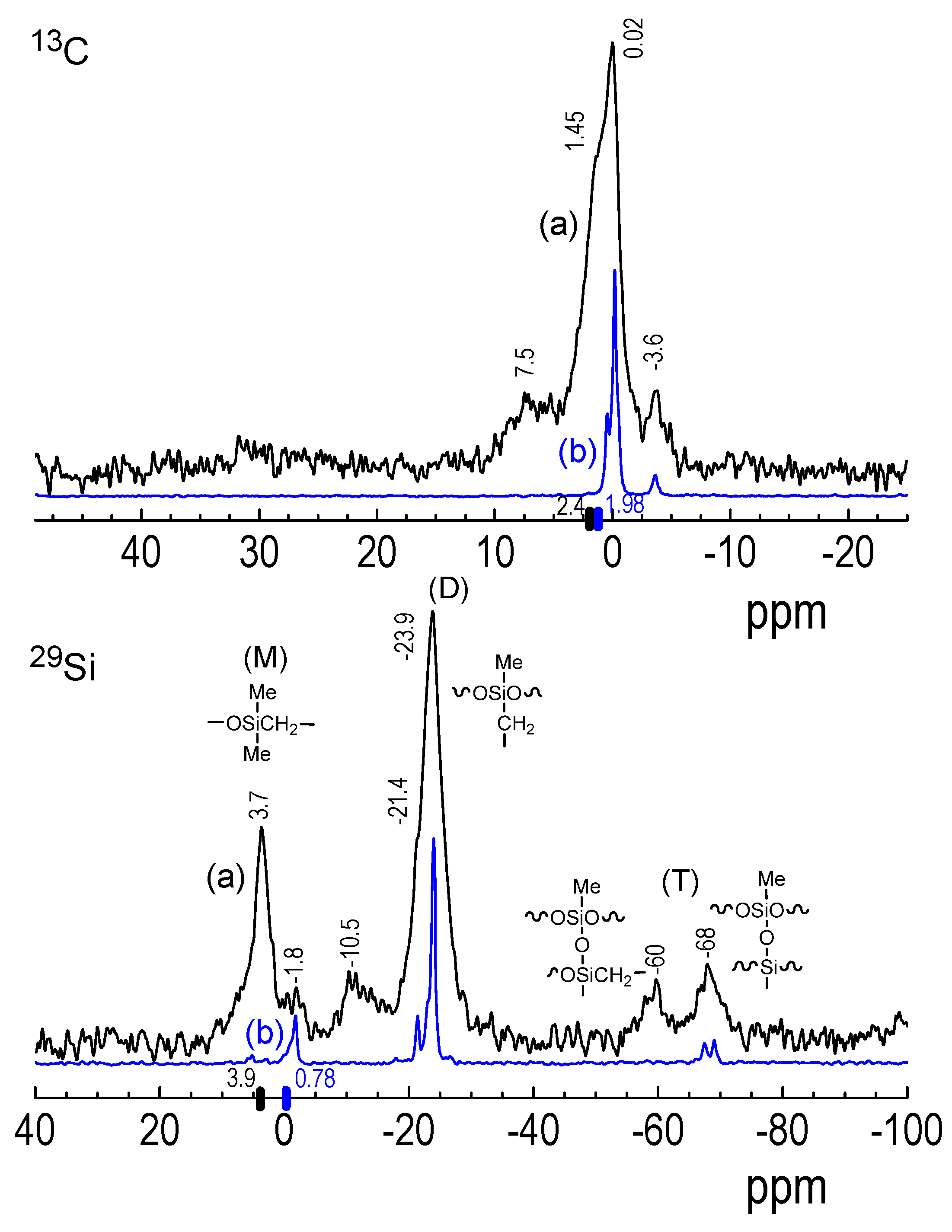
3.3.2. 29Si and 13C NMR Spectroscopy of 2D2 CVD Films and RHP-Treated POBDMS Coatings
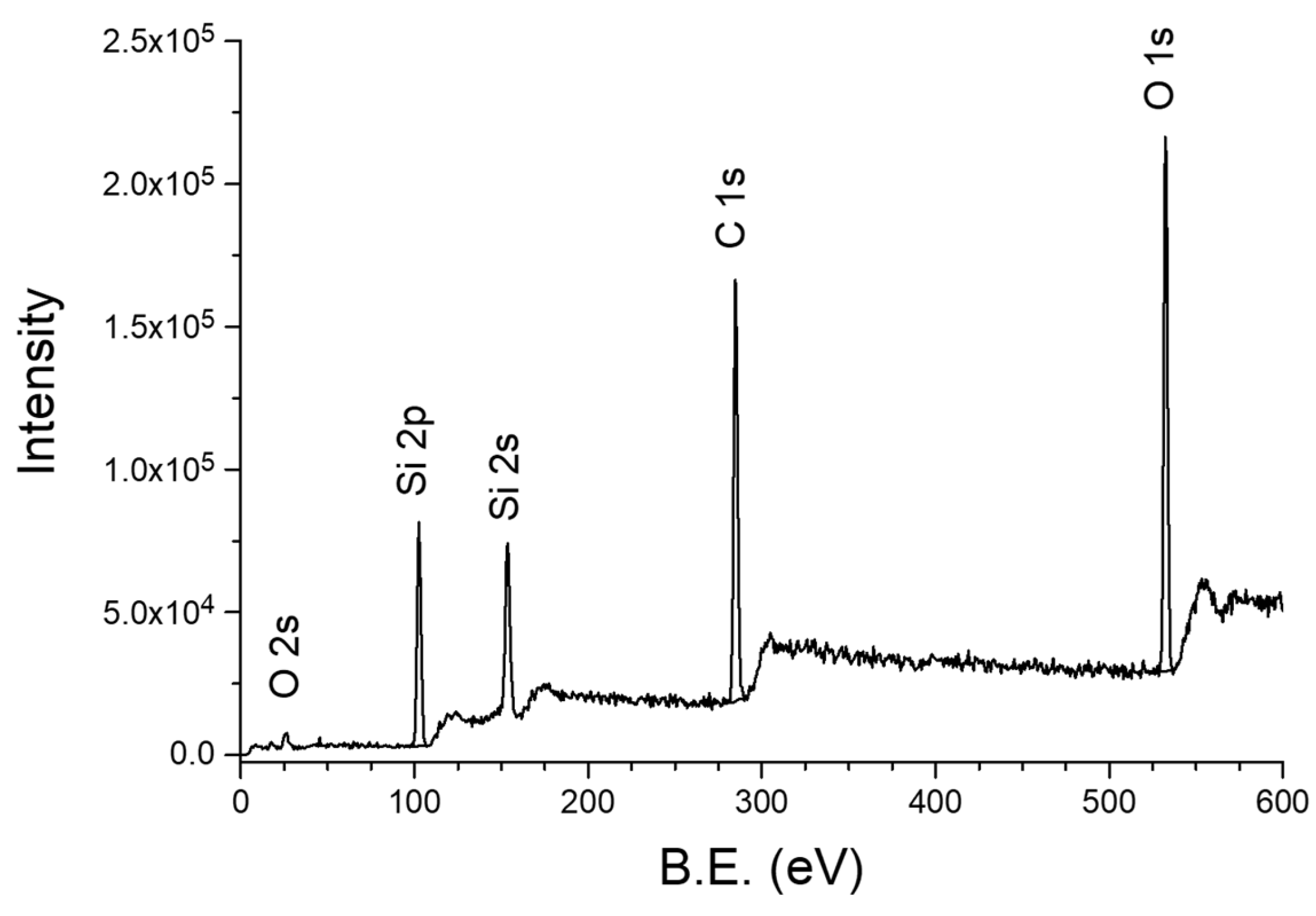
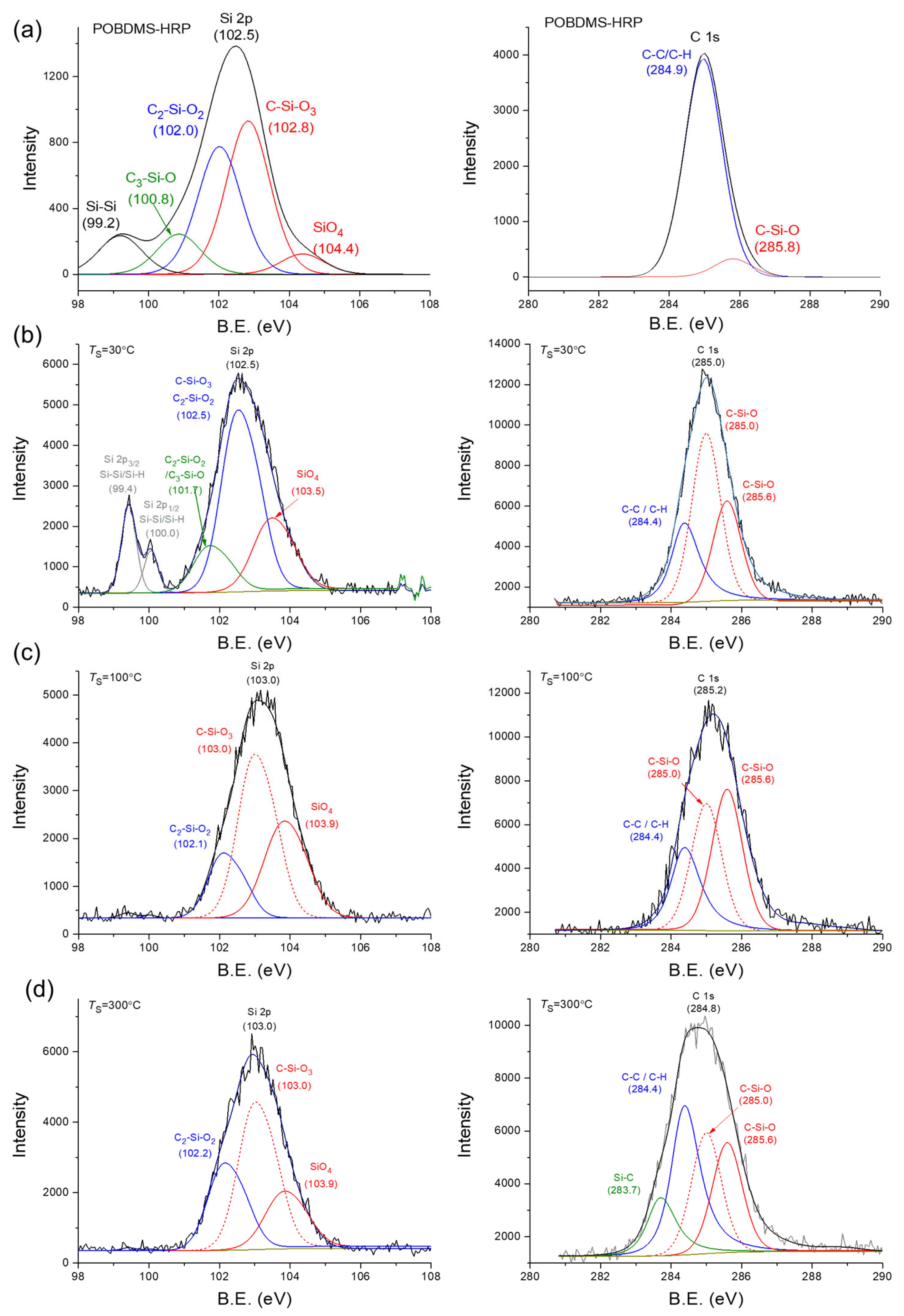
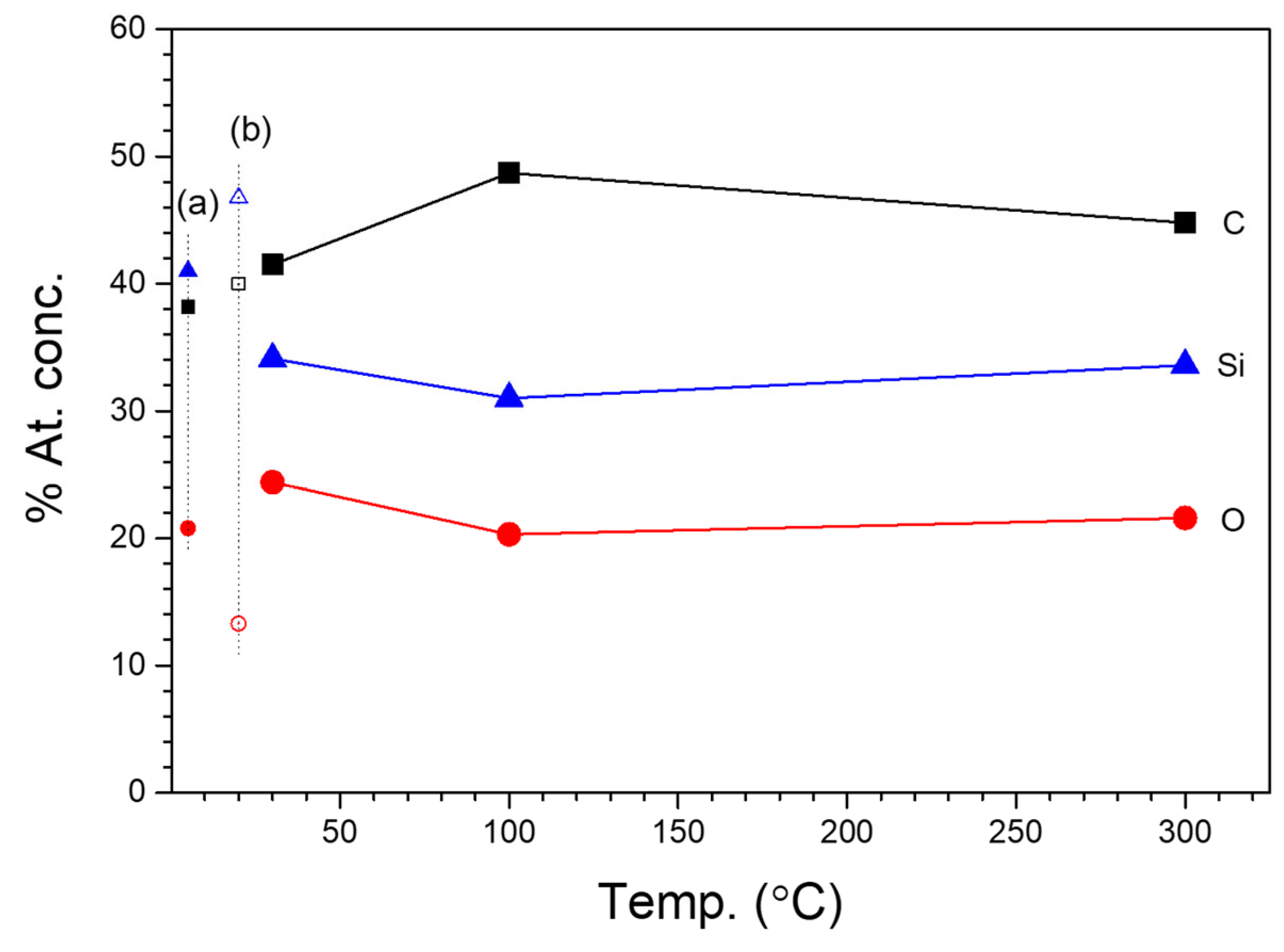
3.3.3. XPS Studies of 2D2-CVD Films and POBDMS RHP-Treated Coatings
3.3.4. Chemical Reactions Involved in the Formation of CVD Layers from the 2D2 Precursor
Activation of Precursor Molecules

Growth Reactions
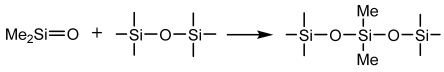
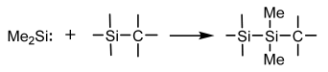
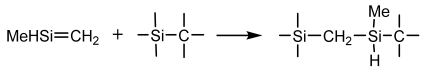

Cross-Linking Reactions
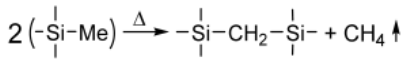
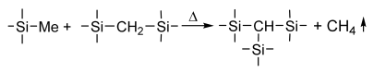
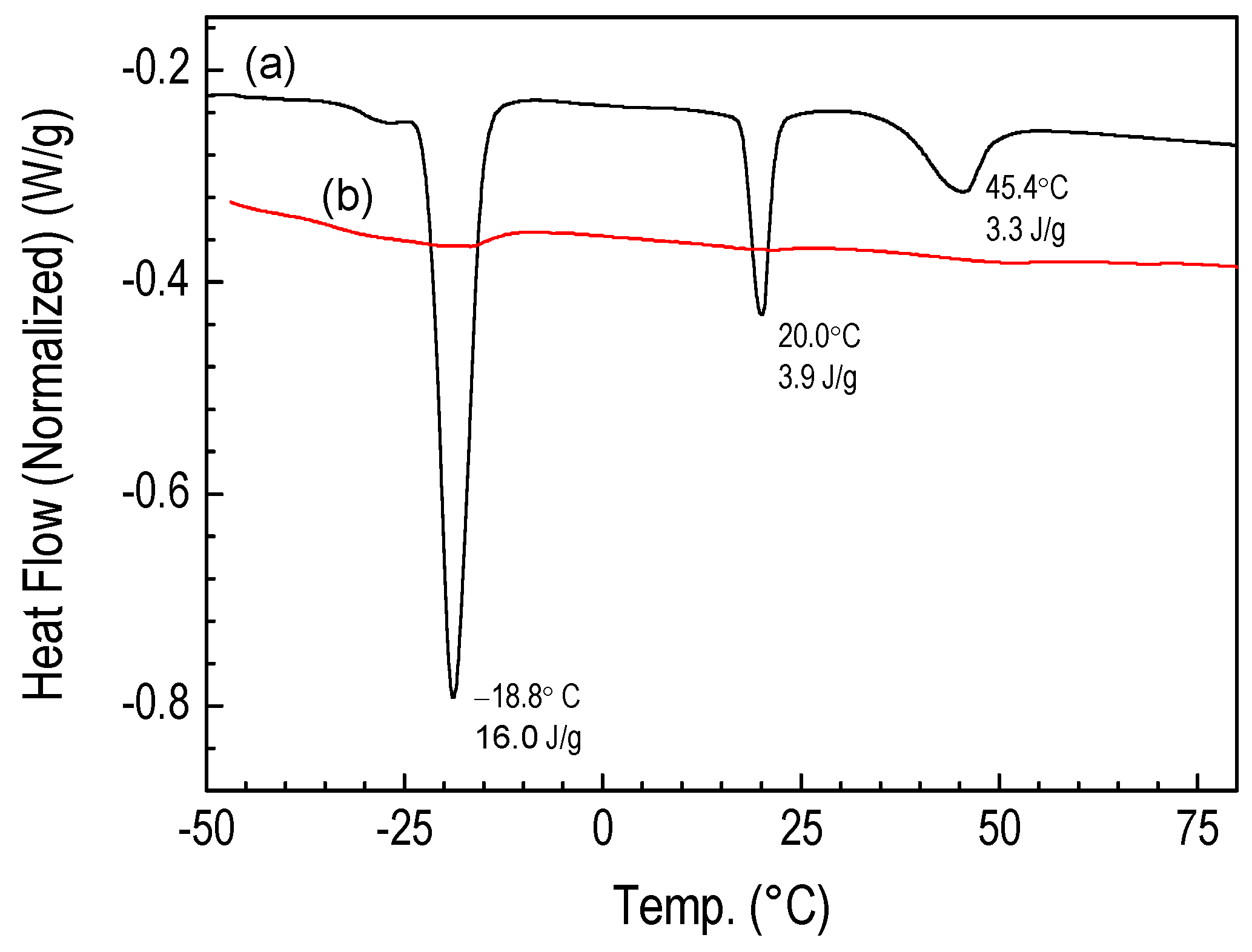
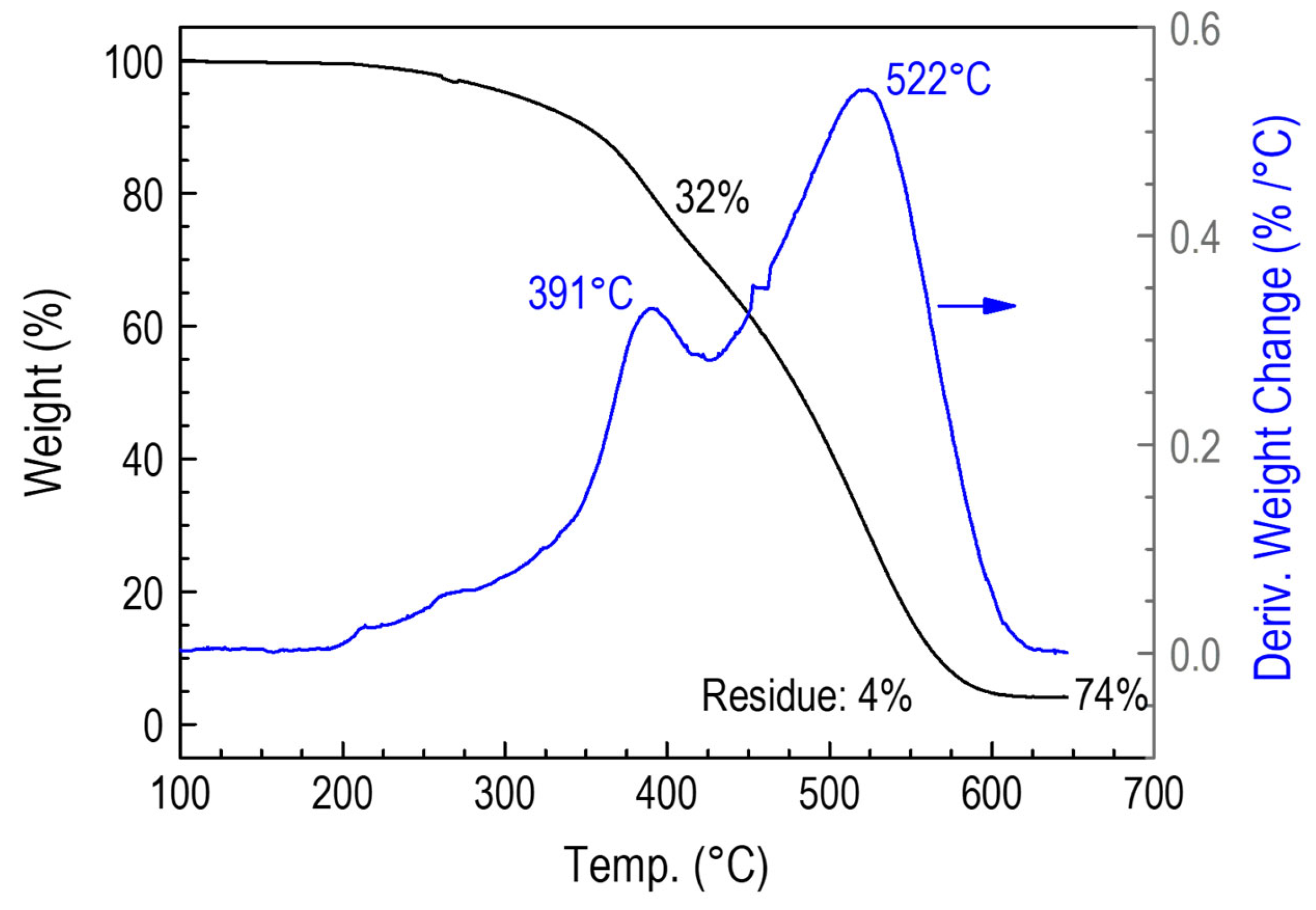
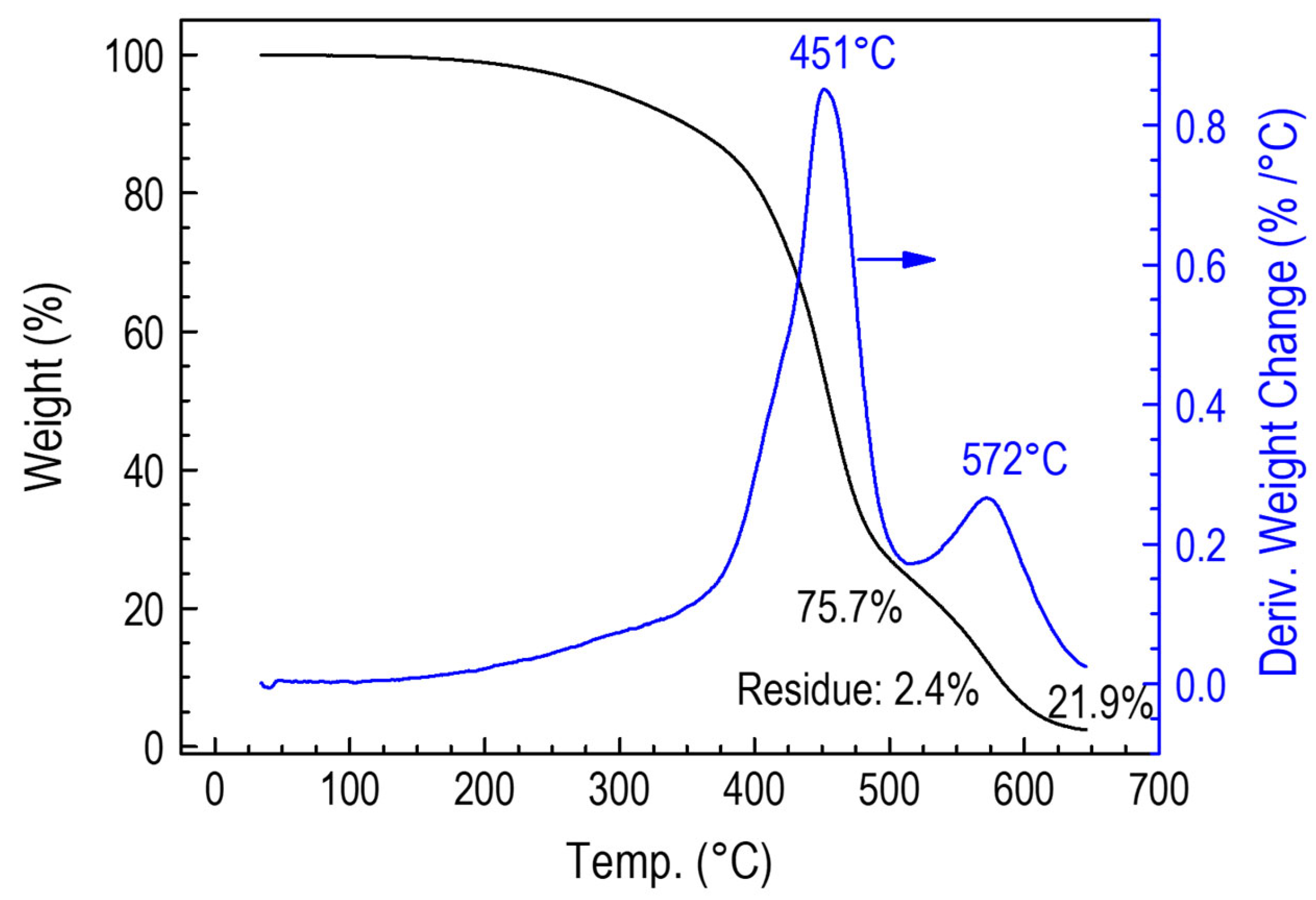
3.3.5. Thermogravimetric Studies of Polymer CVD and RHP-POBDMS Films
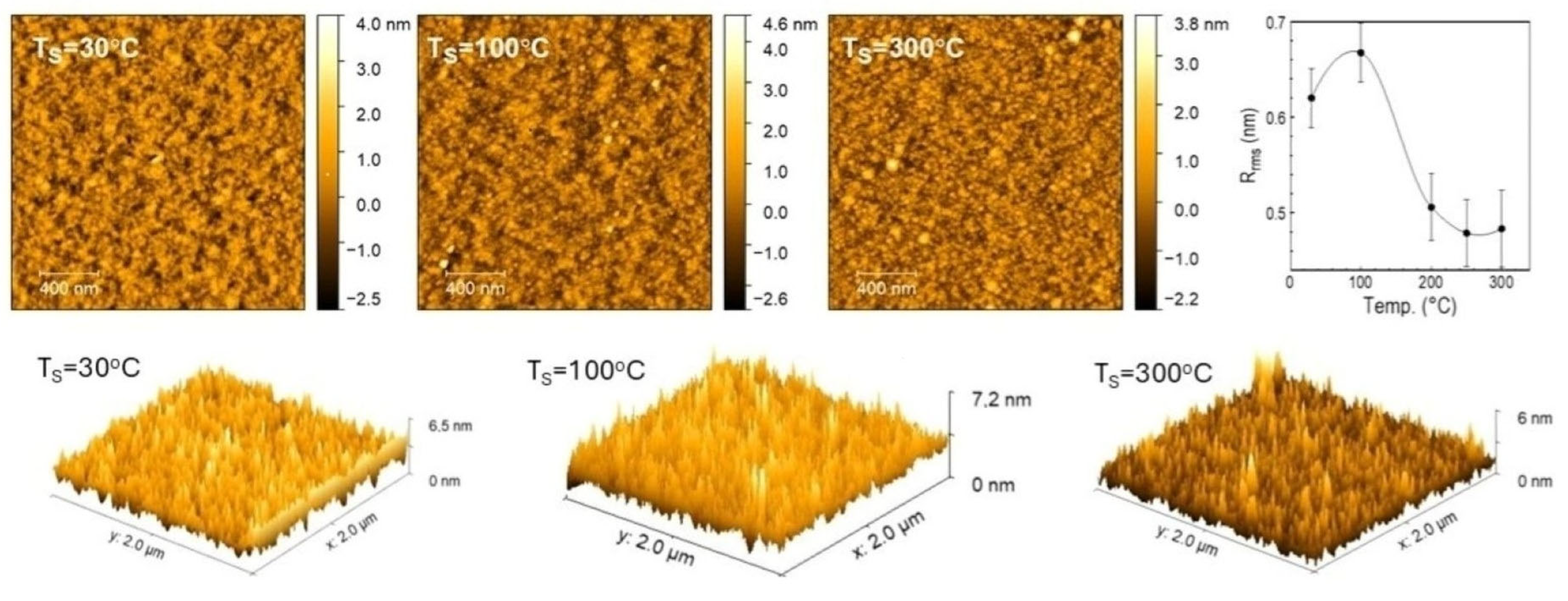
3.3.6. Analysis of Surface Morphology by AFM
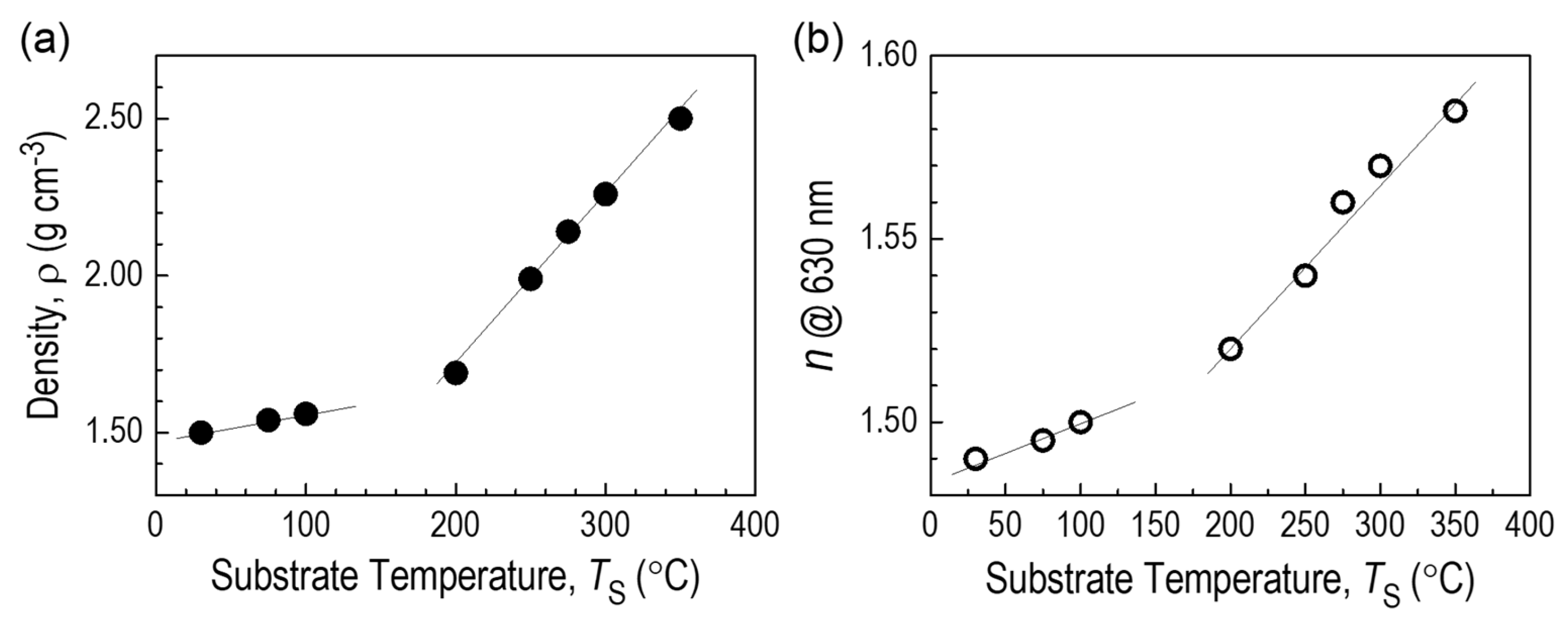
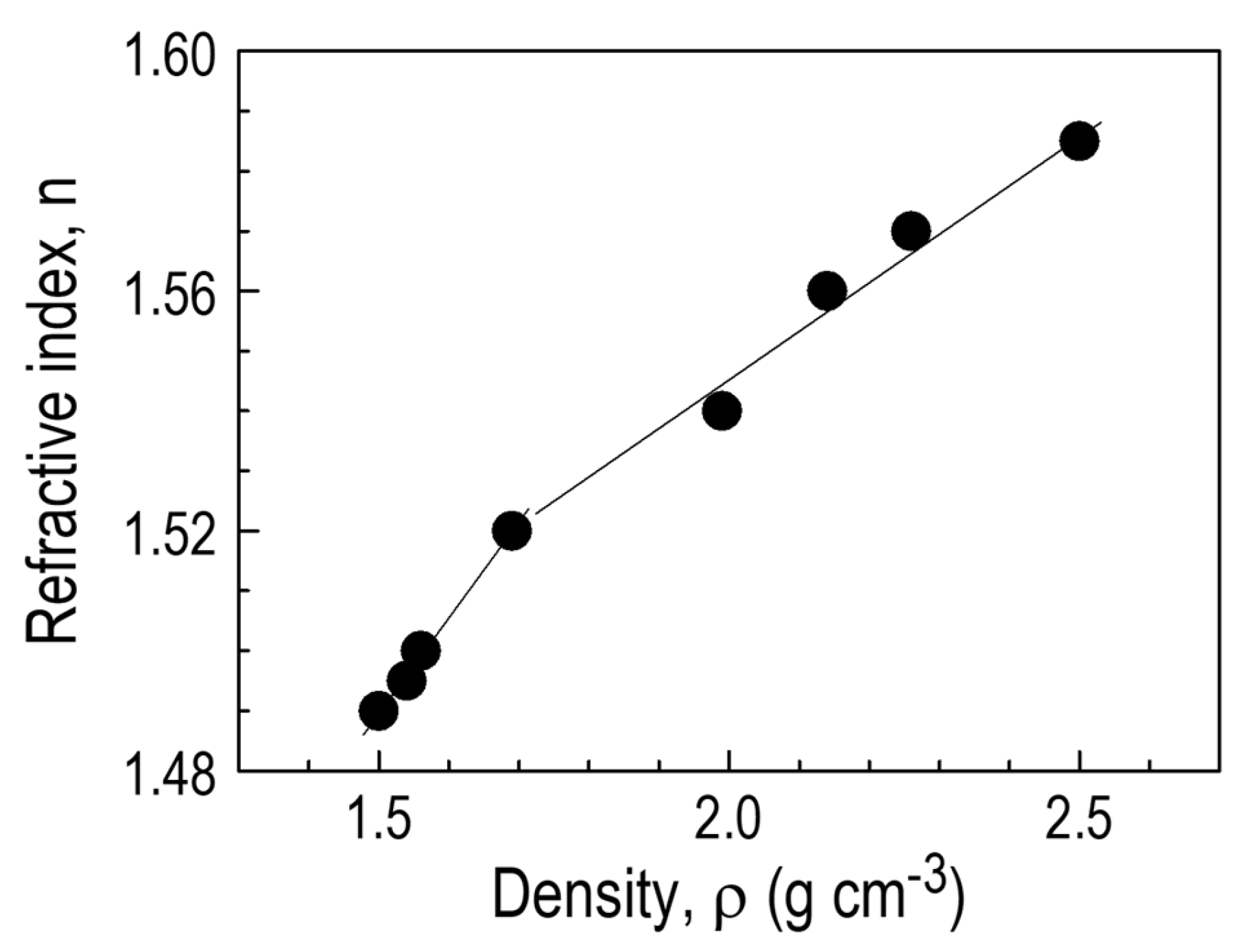
3.3.7. CVD Film Properties—Density and Refractive Index
3.3.8. Photoluminescence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CVD | Chemical Vapor Deposition |
| RHP | Remote Hydrogen Plasma |
| AFM | Atomic Force Microscopy |
| PL | Photoluminescence |
| XPS | X-ray Photoelectron Spectroscopy |
| 2D2 | Octamethyl-1,4-dioxatetrasilacyclohexane |
| POBDMS | Poly(oxybisdimethylsily1ene) |
| RHP-POBDMS | Remote Hydrogen Plasma-treated POBDMS coatings |
| TGA | Thermogravimetric analysis |
| XRD | X-ray diffraction |
References
- Colombo, P.; Mera, G.; Riedel, R.; Sorarù, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Riedel, R.; Mera, G.; Hauser, R.; Klonczynski, A. Silicon-based polymer-derived ceramics: Synthesis properties and applications-a review dedicated to Prof. Dr. Fritz Aldinger on the occasion of his 65th birthday. J. Ceram. Soc. Jpn. 2006, 114, 425–444. [Google Scholar] [CrossRef]
- Schulz, M. Polymer derived ceramics in MEMS/NEMS—A review on production processes and application. Adv. Appl. Ceram. 2009, 108, 454–460. [Google Scholar] [CrossRef]
- Walkiewicz-Pietrzykowska, A.; Uznanski, P.; Wrobel, A.M. Silicon Carbide, Silicon Carbonitride, and Silicon Oxycarbide Thin Films Formed by Remote Hydrogen Microwave Plasma CVD. Curr. Org. Chem. 2017, 21, 2229–2239. [Google Scholar] [CrossRef]
- Szymanowski, H.; Olesko, K.; Kowalski, J.; Fijalkowski, M.; Gazicki-Lipman, M.; Sobczyk-Guzenda, A. Thin SiNC/SiOC Coatings with a Gradient of Refractive Index Deposited from Organosilicon Precursor. Coatings 2020, 10, 794. [Google Scholar] [CrossRef]
- Sujith, R.; Gangadhar, J.; Greenough, M.; Bordia, R.K.; Panda, D.K. A review of silicon oxycarbide ceramics as next generation anode materials for lithium-ion batteries and other electrochemical applications. J. Mater. Chem. A 2023, 11, 20324–20348. [Google Scholar] [CrossRef]
- Brook, M.A. Silicon in Organic, Organometallic, and Polymer Chemistry. Angew. Chem.-Ger. Ed. 2000, 112, 3079. [Google Scholar]
- Lee, S.; Hwang, J.; Lee, J.; Chung, H.; Ryu, D.H.; Yun, H.; Choi, I.G.; Jung, H.; Lee, K.; Yeo, S.; et al. Optimizing low-k SiCOH films deposited by PECVD with a novel C6H16OSi precursor: Impact of oxygen/carbon ratio on film properties. Mater. Chem. Phys. 2025, 334, 130510. [Google Scholar] [CrossRef]
- Park, Y.; Lim, H.; Kwon, S.; Ban, W.; Jang, S.; Jung, D. Ultralow dielectric constant SiCOH films by plasma enhanced chemical vapor deposition of decamethylcyclopentasiloxane and tetrakis(trimethylsilyloxy)silane precursors. Thin Solid Film. 2021, 727, 138680. [Google Scholar] [CrossRef]
- Ermakova, E.; Kosinova, M. Organosilicon compounds as single-source precursors for SiCN films production. J. Organomet. Chem. 2022, 958, 122183. [Google Scholar] [CrossRef]
- Wrobel, A.M.; Uznanski, P.; Walkiewicz-Pietrzykowska, A. Silicon Oxycarbide Films Produced by Remote Microwave Hydrogen Plasma CVD using a Tetramethyldisiloxane Precursor: Growth Kinetics, Structure, Surface Morphology, and Properties. Chem. Vap. Depos. 2015, 21, 307–318. [Google Scholar] [CrossRef]
- Wróbel, A.M.; Walkiewicz-Pietrzykowska, A.; Uznanski, P.; Glebocki, B. Amorphous Hydrogenated Silicon Carbide (a-SiC:H) Coatings Produced by Remote Hydrogen Microwave Plasma CVD from Bis(dimethylsilyl)ethane—A Novel Single-Source Precursor. Chem. Vap. Depos. 2011, 17, 186–190. [Google Scholar] [CrossRef]
- Chojnowski, J.; Cypryk, M.; Kurjata, J. Organic polysilanes interrupted by heteroatoms. Prog. Polym. Sci. 2003, 28, 691–728. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis using SiH Chemistry: The Piers–Rubinsztajn Reaction. Chem. A Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Chojnowski, J.; Kurjata, J. Ring-Opening Polymerization of Octamethyltetrasila-1,4-dioxane, 2D2. 2. Cyclic Oligomer Formation and Mechanism of the Reaction. Macromolecules 1994, 27, 2302–2309. [Google Scholar] [CrossRef]
- Chojnowski, J.; Kurjata, J. Selective Anionic Ring-Opening Polymerization of Permethyltetrasila-1,4-dioxane, 2D2. Transformation of Poly(silaether) in Polysiloxane and Polysilylene. Macromolecules 1995, 28, 2996–2999. [Google Scholar] [CrossRef]
- Pluta, M.; Pakula, T.; Kryszewski, M.; Kurjata, J.; Chojnowski, J. Morphology, phase transitions and viscoelastic properties of poly (oxybisdimethylsilylene). A mesophase in a silicon analogue of a polyether. Macromol. Chem. Phys. 1995, 196, 1607–1623. [Google Scholar] [CrossRef]
- Uznanski, P.; Glebocki, B.; Walkiewicz-Pietrzykowska, A.; Zakrzewska, J.; Wrobel, A.M.; Balcerzak, J.; Tyczkowski, J. Surface modification of silicon oxycarbide films produced by remote hydrogen microwave plasma chemical vapour deposition from tetramethyldisiloxane precursor. Surf. Coat. Technol. 2018, 350, 686–698. [Google Scholar] [CrossRef]
- Wrobel, A.M.; Walkiewicz-Pietrzykowska, A.; Uznanski, P. Remote hydrogen microwave plasma chemical vapor deposition from methylsilane precursors. 2. Surface morphology and properties of deposited a-SiC:H films. Thin Solid Film. 2014, 564, 232–240. [Google Scholar] [CrossRef]
- Wrobel, A.M.; Blaszczyk, I.; Walkiewicz-Pietrzykowska, A.; Tracz, A.; Klemberg-Sapieha, J.E.; Aoki, T.; Hatanaka, Y. Remote hydrogen-nitrogen plasma chemical vapor deposition from a tetramethyldisilazane source. Part 1. Mechanism of the process, structure and surface morphology of deposited amorphous hydrogenated silicon carbonitride films. J. Mater. Chem. 2003, 13, 731–737. [Google Scholar] [CrossRef]
- Niemiec, W.; Szczygieł, P.; Jeleń, P.; Handke, M. IR investigation on silicon oxycarbide structure obtained from precursors with 1:1 silicon to carbon atoms ratio and various carbon atoms distribution. J. Mol. Struct. 2018, 1164, 217–226. [Google Scholar] [CrossRef]
- King, S.W.; Bielefeld, J.; French, M.; Lanford, W.A. Mass and bond density measurements for PECVD a-SiCx:H thin films using Fourier transform-infrared spectroscopy. J. Non-Cryst. Solids 2011, 357, 3602–3615. [Google Scholar] [CrossRef]
- Lin, Y.; Tsui, T.Y.; Vlassak, J.J. Octamethylcyclotetrasiloxane-based, low-permittivity organosilicate coatings: Composition, structure, and polarizability. J. Electrochem. Soc. 2006, 153, F144. [Google Scholar] [CrossRef]
- Lewis, H.G.P.; Casserly, T.B.; Gleason, K.K. Hot-Filament Chemical Vapor Deposition of Organosilicon Thin Films from Hexamethylcyclotrisiloxane and Octamethylcyclotetrasiloxane. J. Electrochem. Soc. 2001, 148, F212. [Google Scholar] [CrossRef]
- Wróbel, A.M.; Walkiewicz-Pietrzykowska, A.; Klemberg-Sapieha, J.E.; Hatanaka, Y.; Aoki, T.; Nakanishi, Y. Remote hydrogen plasma chemical vapor deposition of silicon–carbon thin-film materials from a hexamethyldisilane source: Characterization of the process and the deposits. J. Appl. Polym. Sci. 2002, 86, 1445–1458. [Google Scholar] [CrossRef]
- Launer, P.J.; Arkles, B. Infrared analysis of organosilicon compounds: Spectra-structure correlations. Silicone Compd. Regist. Rev. 2013, 100, 100–103. [Google Scholar]
- Lodhe, M.; Babu, N.; Selvam, A.; Balasubramanian, M. Synthesis and characterization of high ceramic yield polycarbosilane precursor for SiC. J. Adv. Ceram. 2015, 4, 307–311. [Google Scholar] [CrossRef]
- Uznanski, P.; Walkiewicz-Pietrzykowska, A.; Jankowski, K.; Zakrzewska, J.; Wrobel, A.M.; Balcerzak, J.; Tyczkowski, J. Atomic Hydrogen Induced Chemical Vapor Deposition of Silicon Oxycarbide Thin Films Derived from Diethoxymethylsilane Precursor. Appl. Organomet. Chem. 2020, 34, e5674. [Google Scholar] [CrossRef]
- Roualdes, S.; Berjoan, R.; Durand, J. 29Si NMR and Si2p XPS correlation in polysiloxane membranes prepared by plasma enhanced chemical vapor deposition. Sep. Purif. Technol. 2001, 25, 391–397. [Google Scholar] [CrossRef]
- Hung, I.; Ionescu, E.; Sen, J.; Gan, Z.; Sen, S. Structure and Connectivity in an Amorphous Silicon Oxycarbide Polymer-Derived Ceramic: Results from 2D 29Si NMR Spectroscopy. J. Phys. Chem. C 2021, 125, 4777–4784. [Google Scholar] [CrossRef]
- Lee, J.; Jang, W.; Kim, H.; Shin, S.; Kweon, Y.; Lee, K.; Jeon, H. Characteristics of low-κ SiOC films deposited via atomic layer deposition. Thin Solid Film. 2018, 645, 334–339. [Google Scholar] [CrossRef]
- Shi, Y.; Li, X.; Tong, L.; Toukabri, R.; Eustergerling, B. Decomposition of hexamethyldisilane on a hot tungsten filament and gas-phase reactions in a hot-wire chemical vapor deposition reactor. Phys. Chem. Chem. Phys. 2008, 10, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, K.D.; Doren, D.J. Mechanistic insight into gas-phase reactions of hydrogen atom with disilane and hydrogen atom etching of silicon surfaces. J. Am. Chem. Soc. 1993, 115, 3731–3738. [Google Scholar] [CrossRef]
- O’Neal, H.E.; Ring, M.A. Mechanism of the thermal decomposition of dimethylsilane at atmospheric pressures in the gas phase. Organometallics 1988, 7, 1017–1025. [Google Scholar] [CrossRef]
- Birot, M.; Pillot, J.-P.; Dunogues, J. Comprehensive Chemistry of Polycarbosilanes, Polysilazanes, and Polycarbosilazanes as Precursors of Ceramics. Chem. Rev. 1995, 95, 1443–1477. [Google Scholar] [CrossRef]
- Wrobel, A.M.; Walkiewicz-Pietrzykowska, A. Mechanism of the initiation step in atomic hydrogen-induced CVD of amorphous hydrogenated silicon-carbon films from single-source precursors. Chem. Vap. Depos. 1998, 4, 133–141. [Google Scholar] [CrossRef]
- Raabe, G.; Michl, J. Multiple bonding to silicon. Chem. Rev. 1985, 85, 419–509. [Google Scholar] [CrossRef]
- Ma, J.; Shi, L.; Shi, Y.; Luo, S.; Xu, J. Pyrolysis of polymethylsilsesquioxane. J. Appl. Polym. Sci. 2002, 85, 1077–1086. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Chernyavskaya, N.A.; Antipin, M.Y.; Lysenko, K.A.; Chernyavskii, A.I. Molecular and Crystal Structure of Octamethyl-1,4-dioxacyclohexasilane. Chem. Heterocycl. Compd. 2005, 41, 536–541. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Measurement |
|---|---|
| 29Si and 13C CP/MAS NMR | Chemical structure |
| Fourier transform infrared (FTIR) | Substituent groups |
| X-ray Photoelectron Spectroscopy (XPS) | Atomic concentration |
| X-ray diffraction (XRD) | Crystallographic structure |
| Spectroscopic ellipsometry | Thickness and refractive index |
| Atomic force microscopy (AFM) | Particle size and roughness |
| Photoluminescence (PL) | Photoluminescence excitation and emission |
| Thermogravimetric analysis (TGA) | Thermal stability, decomposition, and compositional changes |
| Band Assignment * | Monomer | CVD LayerTS = 30 °C | CVD LayerTS = 350 °C | POBDMS | POBDMS After RHP |
|---|---|---|---|---|---|
| νas(C-H) in CH3 | 2956 | 2957 | - | 2953 | 2958 |
| νs(C-H) in CH3 | 2894, 2795 | 2901, 2796 | - | 2894, 2793 | 2897 |
| ν(Si-H) | - | 2126 | - | 2122 | |
| δas(CH3) in Si-CH3 | 1401 | 1407 | - | 1403 | 1404 |
| δ(CH2) in Si-CH2-Si | - | 1355 | - | ||
| δs(CH3) in Si-CH3 | 1248 | 1259 | 1264 | 1250 | 1260 |
| νas(SiOSi) | 1064, 1026 sh Cyclic siloxanes have only 1 band | - | - | 1056 sh, 1025 D type dimethylsiloxane linear chains | - |
| νas(SiOSi), chain or small cycle, ν(Si-C-Si) in (Si-CH2-Si) | - | 1056 sh, 1027 | 1133sh, νas(Si-O-Si) cavity 1002 νas(SiOC) | - | 1056, 1033 |
| δ(Si-H) | - | 909 | - | - | 910 |
| ρ(CH3) in Si(CH3)2 | 847sh | 832 | 812sh | 853, 832 | 853, 832 |
| ρ(CH3) in the Si-Si group, ν(SiC) | 806 | 798 | 792, 733sh ν(SiC) carbide | 815 | 800 |
| ρ(CH3) in Si-CH3 | 770 | 771 | - | 768 | 768 |
| ν(SiC) in ((CH3)2Si)2 group | 714, 679 | 728, 683 | - | 681 | 686 |
| 611 Si-Si in Crystal | - | - | - | 644 | - |
| νs(SiSiO) Si-Si-O in the ring | 542 | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walkiewicz-Pietrzykowska, A.; Jankowski, K.; Kurjata, J.; Dolot, R.; Brzozowski, R.; Zakrzewska, J.; Uznanski, P. Silicon Oxycarbide Thin Films Produced by Hydrogen-Induced CVD Process from Cyclic Dioxa-Tetrasilacyclohexane. Materials 2025, 18, 2911. https://doi.org/10.3390/ma18122911
Walkiewicz-Pietrzykowska A, Jankowski K, Kurjata J, Dolot R, Brzozowski R, Zakrzewska J, Uznanski P. Silicon Oxycarbide Thin Films Produced by Hydrogen-Induced CVD Process from Cyclic Dioxa-Tetrasilacyclohexane. Materials. 2025; 18(12):2911. https://doi.org/10.3390/ma18122911
Chicago/Turabian StyleWalkiewicz-Pietrzykowska, Agnieszka, Krzysztof Jankowski, Jan Kurjata, Rafał Dolot, Romuald Brzozowski, Joanna Zakrzewska, and Paweł Uznanski. 2025. "Silicon Oxycarbide Thin Films Produced by Hydrogen-Induced CVD Process from Cyclic Dioxa-Tetrasilacyclohexane" Materials 18, no. 12: 2911. https://doi.org/10.3390/ma18122911
APA StyleWalkiewicz-Pietrzykowska, A., Jankowski, K., Kurjata, J., Dolot, R., Brzozowski, R., Zakrzewska, J., & Uznanski, P. (2025). Silicon Oxycarbide Thin Films Produced by Hydrogen-Induced CVD Process from Cyclic Dioxa-Tetrasilacyclohexane. Materials, 18(12), 2911. https://doi.org/10.3390/ma18122911