Activated Carbon/Transition Metal (Ni, In, Cu) Hexacyanoferrate Nanocomposites for Cesium Adsorption



 ,
, 
Abstract
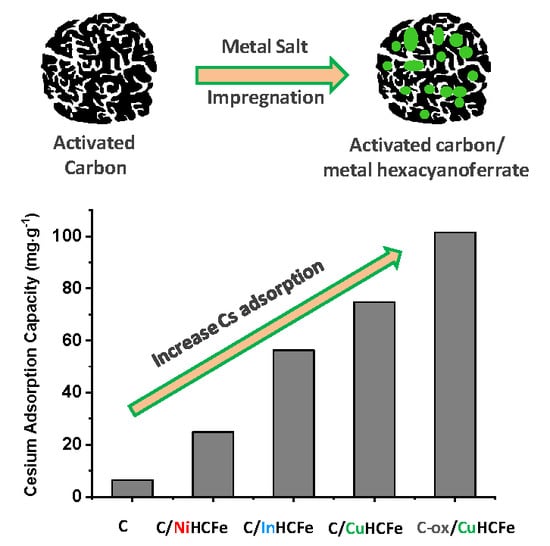
1. Introduction
2. Materials and Methods
2.1. Materials Synthesis
2.2. Materials Characterization
2.3. Cesium Adsorption Tests
3. Results and Discussion
3.1. Influence of the Transition Metal Type
3.2. Influence of the Carbon Surface Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alinaghizadeh, H.; Wålinder, R.; Vingård, E.; Tondel, M. Total cancer incidence in relation to 137 Cs fallout in the most contaminated counties in Sweden after the Chernobyl nuclear power plant accident: A register-based study. BJM Open 2016, 6, 1–8. [Google Scholar]
- Liu, X.; Chen, G.R.; Lee, D.J.; Kawamoto, T.; Tanaka, H.; Chen, M.L.; Luo, Y.K. Adsorption removal of cesium from drinking waters: A mini review on use of biosorbents and other adsorbents. Bioresour. Technol. 2014, 160, 142–149. [Google Scholar] [CrossRef]
- Zhang, H.; Kim, Y.; Hunter, T.; Brown, A.; Leeb, J.; Harbottle, D. Organically modified clay with potassium copper hexacyanoferrate for enhanced Cs+ adsorption capacity and selective recovery by flotation. J. Mater. Chem. A 2017, 5, 15130–15143. [Google Scholar]
- Vanderheyden, S.R.; Van Ammel, R.; Sobiech-Matura, K.; Vanreppelen, K.; Schreurs, S.; Schroeyers, W.; Yperman, J.; Carleer, R. Adsorption of cesium on different types of activated carbon. J. Radioanal. Nucl. Chem. 2016, 310, 301–310. [Google Scholar]
- Koshima, H.; Onishi, H. Adsorption of metal ions on activated carbon from aqueous solutions at pH 1–13. Talanta 1986, 33, 391–395. [Google Scholar] [PubMed]
- Song, K.; Kim, H.; Lee, H.; Park, H.; Lee, K. Adsorption characteristics of radiotoxic cesium and iodine from low-level liquid wastes. J. Radioanal. Nucl. Chem. 1997, 223, 199–205. [Google Scholar] [CrossRef]
- Hassan, H.; Attallah, M.; Yakout, S. Sorption characteristics of an economical sorbent material used for removal radioisotopes of cesium and europium. J. Radioanal. Nucl. Chem. 2010, 286, 17–26. [Google Scholar]
- Gad, H.; Elsanafini, H.; Ali, M.; Lasheen, Y.; Abdelwahed, M. Factors affecting the sorption behavior of Cs+ and Sr2+ using biosorbent material. Russ. J. Appl. Chem. 2016, 89, 988–999. [Google Scholar]
- Zakla-El, T.; Yakout, S.; Rizk, M.; Lasheen, Y.; Gad, H. Removal of Cobalt-60 and Caesium-134 Ions from Contaminated Solutions by Sorption Using Activated Carbon. Adsorpt. Sci. Technol. 2011, 29, 331–344. [Google Scholar] [CrossRef]
- Caccin, M.; Giacobbo, F.; da Ros, M.; Besozzi, M.; Mariani, M. Adsorption of uranium, cesium and strontium onto coconut shell activated carbon. J. Radioanal. Nucl. Chem. 2013, 297, 9–18. [Google Scholar] [CrossRef]
- Alarifi, A.; Hanafi, H. Adsorption of cesium, thallium, strontium and cobalt radionuclides using activated carbon. Asian J. Chem. 2011, 23, 111–115. [Google Scholar]
- Loos-Neskovic, C.; Ayrault, S.; Badillo, V.; Jimenez, B.; Garnier, E.; Fedoroff, M.; Jones, D.J.; Merinov, B. Structure of copper-potassium hexacyanoferrate (II) and sorption mechanisms of cesium. J. Solid State Chem. 2004, 177, 1817–1828. [Google Scholar]
- Ayrault, S.; Jimenez, B.; Garnier, E.; Fedoroff, M.; Jones, D.; Loos-Neskovic, C. Sorption Mechanisms of Cesium on CuII2FeII(CN)6 and CuII3[FeIII(CN)6]2 Hexacyanoferrates and Their Relation to the Crystalline Structure. J. Solid State Chem. 1998, 141, 475–485. [Google Scholar] [CrossRef]
- Mimura, H.; Kageymana, N.; Akiba, K.; Yoneya, M.; Miyamoto, Y. Ion-exchange properties of potassium nickel hexacyanoferrate (II) compounds. Solvent Extr. Ion Exch. 1998, 16, 1013–1031. [Google Scholar]
- Ismail, I.; El-Sourougy, M.; Moneim, N.; Aly, H. Equilibrium and kinetic studies of the sorption of cesium by potassium nickel hexacyanoferrate complex. J. Radioanal. Nucl. Chem. 1999, 240, 59–67. [Google Scholar] [CrossRef]
- Ismail, I.; El-Sourougy, M.; Moneim, N.; Aly, H. Preparation, characterization, and utilization of potassium nickel hexacyanoferrate for the separation of cesium and cobalt from contaminated waste water. J. Radioanal. Nucl. Chem. 1998, 237, 97–103. [Google Scholar]
- Grandjean, A.; Delchet, C.; Causse, J.; Barré, Y.; Guari, Y.; Larionova, J. Effect of the chemical nature of different transition metal ferrocyanides to entrap Cs. J. Radioanal. Nucl. Chem. 2016, 307, 427–436. [Google Scholar] [CrossRef]
- Vincent, T.; Vincent, C.; Guibal, E. Immobilization of Metal Hexacyanoferrate Ion-Exchangers for the Synthesis of Metal Ion Sorbents-A Mini-Review. Molecules 2015, 20, 20582–20613. [Google Scholar] [CrossRef]
- Vipin, A.K.; Fugetsu, B.; Sakata, I.; Isogai, A.; Endo, M.; Li, M.; Dresselhaus, M.S. Cellulose nanofiber backboned Prussian blue nanoparticles as powerful adsorbents for the selective elimination of radioactive cesium. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- Kawamura, F.; Motojima, K. Using copper hexacyanoferrate (II) impregnated zeolite for cesium removal from radioactive liquid waste. Nucl. Technol. 1982, 58, 242–247. [Google Scholar]
- Mimura, H.; Kimura, M.; Akiba, K.; Onodera, Y. Separation of Cesium and Strontium by Potassium Nickel: Hexacyanoferrate(II)-Loaded Zeolite A. J. Nucl. Sci. Technol. 1999, 36, 307–310. [Google Scholar] [CrossRef][Green Version]
- Sangvanich, T.; Sukwarotwat, V.; Wiacek, R.J.; Grudzien, R.M.; Fryxell, G.E.; Addleman, R.S.; Timchalk, C.; Yantasee, W. Selective capture of cesium and thallium from natural waters and simulated wastes with copper ferrocyanide functionalized mesoporous silica. J. Hazard. Mater. 2010, 182, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Chau, L.-K.; Hu, W.-P.; Wang, C.-Y.; Liao, J.-H. Nickel hexacyanoferrate multilayers on functionalized mesoporous silica supports for selective sorption and sensing of cesium. Micro Mesoporous Mater. 2008, 109, 505–512. [Google Scholar] [CrossRef]
- Van Minh, N.; Vo, V.; Lee, H.; Kim, J.; Kim, S. Synthesis and characterization of Co-Fe Prussian blue nanoparticles within MCM-41. Mater. Res. Bull. 2009, 44, 78–81. [Google Scholar]
- Shiozaki, K.; Shigemoto, N. Comparative study of Cs-uptake performance by potassium iron(III) hexacyanoferrate(II) supported on wood chips and activated carbon. J. Chem. Eng. Jpn. 2013, 46, 601–608. [Google Scholar] [CrossRef]
- Hong, J.-Y.; Oh, W.-K.; Shin, K.-Y.; Kwon, O.; Son, S.; Jang, J. Spatially controlled carbon sponge for targeting internalized radioactive materials in human body. Biomaterials 2012, 33, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Kawatake, K.; Shigemoto, N. Preparation of potassium iron(III) hexacyanoferrate(II) supported on activated carbon and Cs uptake performance of the adsorbent. J. Nucl. Sci. Technol. 2012, 49, 1048–1056. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Feng, M.; Liu, C.; Zhao, Y.; Li, S.; Wang, H.; Yan, L.; Tian, G.; Li, S. Supporting of potassium copper hexacyanoferrate on porous activated carbon substrate for cesium separation. Sep. Sci. Technol. 2009, 44, 4023–4035. [Google Scholar] [CrossRef]
- Dashtinejad, M.; Samadfam, M.; Fasihi, J.; Fumeshkenar, F.G.; Sepehrian, H. Synthesis, characterization, and cesium sorption performance of potassium nickel hexacyanoferrate-loaded granular activated carbon. Part. Part. Synth. Tehnol. 2014, 32, 348–354. [Google Scholar] [CrossRef]
- Lalhriatpuia, C.; Tiwari, D.; Lee, S.M. Immobilized nickel hexacyanoferrate on activated carbons for efficient attenuation of radio toxic Cs(I) from aqueous solutions. Appl. Surf. Sci. 2014, 321, 275–282. [Google Scholar]
- Jeerage, K.; Schwartz, D. Characterization of cathodically deposited nickel hexacyanoferrate for electrochemically switched ion exchange. Sep. Sci. Technol. 2000, 35, 2375–2392. [Google Scholar] [CrossRef]
- Jagiello, J.; Olivier, J. 2D-NLDFT adsorption models for carbon slit-shaped pores with surface energetical heterogeneity and geometrical corrugation. Carbon 2013, 55, 70–80. [Google Scholar] [CrossRef]
- Caussin, P.; Nusinovici, J.; Beard, D.W. Using digitized X-ray powder diffraction scans as input for a new PC-AT Search/match program. Adv. X-ray Anal. 1988, 31, 423–430. [Google Scholar]
- Chen, L.; Shao, H.; Zhou, X.; Liu, G.; Jiang, J.; Liu, Z. Water-mediated cation intercalation of open-framework indium hexacyanoferrate with high voltage and fast kinetics. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Gražulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.F.; Quirós, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database—An open-access collection of crystal structures. J. Appl. Cryst. 2009, 42, 726–729. [Google Scholar] [CrossRef]
- Roisnel, T.; Rodriguez-Carvajal, J. WinPLOTR: A Windows tool for powder diffraction patterns analysis. Materials Science Forum. In Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), Barcelona, Spain, 20–23 May 2000; Delhez, R., Mittenmeijer, E.J., Eds.; Transtec Publications: Wichita, KS, USA, 2000; pp. 118–123. [Google Scholar]
- Kourhoumelis, N. PowDLL: a reusable. NET component for interconverting powder diffraction data: Recent developments. Powder Differ. 2013, 28, 137–148. [Google Scholar]
- Scherrer, P. Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. In Kolloidchemie Ein Lehrbuch; Springer: Berlin/Heidelberg, Germany, 1918; Volume 2, pp. 98–100. [Google Scholar]
- Vincent, T.; Vincent, C.; Barré, Y.; Guari, Y.; le Saoutd, G.; Guibal, E. Immobilization of metal hexacyanoferrates in chitin beads for cesium sorption: Synthesis and characterization. J. Mater. Chem. A 2014, 2, 10021. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Orfao, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Khandaker, S.; Toyahara, Y.; Kamida, S.; Kuba, T. Adsorptive removal of cesium from aqueous solution using oxidized bamboo charcoal. Water Resour. 2018, 19, 35–46. [Google Scholar] [CrossRef]
- Yavari, R.; Huang, Y.; Ahmadi, S. Adsorption of cesium (I) from aqueous solution using oxidized multiwall carbon nanotubes. J. Radioanal. Nucl. Chem. 2011, 287, 393–401. [Google Scholar] [CrossRef]
- Li, N.; Xu, S.; Yuan, J.; Miao, J.; Niu, L.; Song, J. In site formation and growth of Prussian blue nanoparticles anchored to multiwalled carbon nanotubes with poly(4-vinylpyridine) linker by layer-by-layer assembly. Mater. Chem. Phys. 2012, 133, 726–734. [Google Scholar] [CrossRef]
- Draouil, H.; Alvarez, L.; Causse, J.; Flaud, V.; Zaibi, M.A.; Bantignies, J.L.; Oueslati, M.; Cambedouzou, J. Copper hexacyanoferrate functionalized single-walled carbon nanotubes for selective cesium extraction. New J. Chem. 2017, 41, 7705–7713. [Google Scholar]
- Jang, J.; Lee, D. Enhanced adsorption of cesium on PVA-alginate encapsulated Prussian blue-graphene oxide hydrogel beads in a fixed-bed column system. Bioresour. Technol. 2016, 218, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Minami, N.; Tanaka, H.; Sue, K.; Minami, K.; Parajuli, D.; Lee, K.M.; Ohkoshi, S.I.; Kurihara, M.; Kawamoto, T. Efficient synthesis of size-controlled open-framework nanoparticles fabricated with a micro-mixer: Route to the improvement of Cs adsorption performance. Green Chem. 2015, 17, 4228–4233. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, Y.; Kim, S.; Harbottle, D.; Lee, J. Solvent-assisted synthesis of potassium copper hexacyanoferrate embedded 3D-interconnected porous hydrogel for highly selective and rapid cesium ion removal. J. Environ. Chem. Eng. 2017, 5, 975–986. [Google Scholar]
- Han, F.; Zhang, G.-H.; Gu, P. Adsorption kinetics and equilibrium modeling of cesium on copper ferrocyanide. J. Radioanal. Nucl. Chem. 2013, 295, 369–377. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
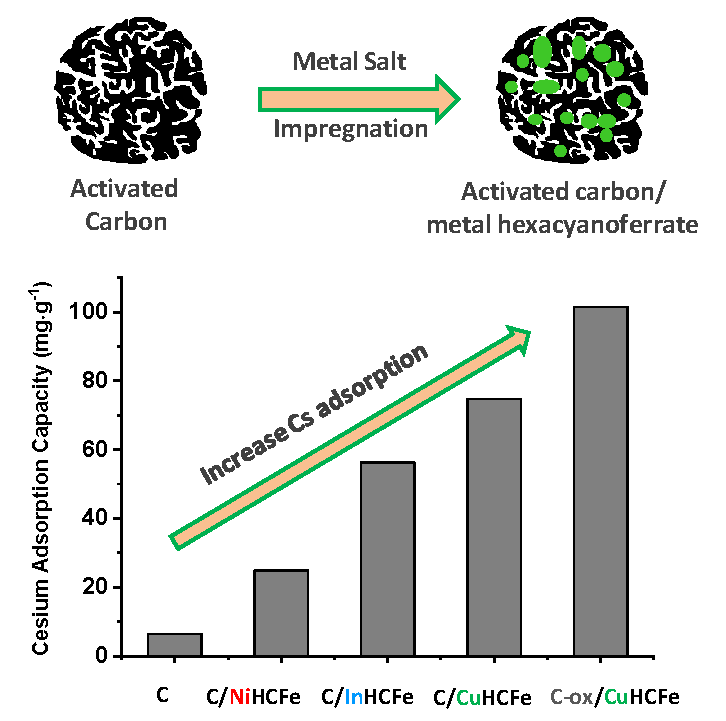
| Material | SBET (m2·g−1) | Vtotal pores (cm3·g−1) | Vmicropores (cm3·g−1) | Vmesopores (cm3·g−1) | TGA Residue (wt%) | Cs Capacity (mg·g−1) |
|---|---|---|---|---|---|---|
| C | 1643 | 0.87 | 0.49 | 0.38 | 6.9 | 6.4 |
| C/NiHCFe | 1799 | 0.85 | 0.55 | 0.30 | 8.3 | 24.9 |
| C/InHCFe | 1342 | 0.65 | 0.41 | 0.24 | 19.5 | 56.3 |
| C/CuHCFe | 1450 | 0.74 | 0.43 | 0.31 | 9.2 | 74.7 |
| C-ox | 573 | 0.31 | 0.21 | 0.10 | 4.2 | 46.9 |
| C-ox/CuHCFe | 246 | 0.12 | 0.09 | 0.03 | 13.6 | 101.5 |
| Materials | C1s | O1s | K2p | N1s | P2p | Cu2p3/2 | Fe2p3/2 | Cs3d |
|---|---|---|---|---|---|---|---|---|
| C | 85.9 | 12.9 | - | - | 1.2 | - | - | - |
| C-ox | 73.8 | 23.4 | - | 2.3 | 0.2 | - | - | - |
| C/CuHCFe | 82.9 | 13.4 | 1.2 | 1.1 | 1.0 | 0.24 | 0.15 | - |
| C-ox/CuHCFe | 68.5 | 17.8 | 3.2 | 7.6 | - | 1.7 | 1.05 | - |
| C/CuHCFe-Cs | 84.5 | 12.6 | - | 1.0 | 0.7 | 0.12 | 0.15 | 0.91 |
| C-ox-Cs | 75.4 | 21.5 | - | 1.9 | - | - | - | 1.16 |
| Adsorbent Type | Adsorbent Name | Sorption Capacity (mg·g−1) | Reference |
|---|---|---|---|
| Activated Carbon | 6 | [28] | |
| Carbon | Oxidized MWCNTs | 12.75 | [42] |
| Oxidized bamboo charcoal | 53.5 | [41] | |
| C-ox | 46.9 | This study | |
| NiHCFe-AC(R) | 31.2 | [30] | |
| AC/KHCFe | 1.38 | [27] | |
| Carbon/MHCFe | AC/CuHCFe | 61.2 | [28] |
| AC/KNiCFe | 163.9 | [29] | |
| C-ox/CuHCFe | 101 | This study | |
| MWCNTs-P4VB-CuHCFe | 150 | [43] | |
| Carbon/Polymer/HCFe | SWCNTs-PRG-CuHCFe | 240 | [44] |
| Graphene Oxide-PVA-CuHCFe | 164.5 | [45] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiener, J.; Limousy, L.; Jeguirim, M.; Le Meins, J.-M.; Hajjar-Garreau, S.; Bigoin, G.; Ghimbeu, C.M. Activated Carbon/Transition Metal (Ni, In, Cu) Hexacyanoferrate Nanocomposites for Cesium Adsorption. Materials 2019, 12, 1253. https://doi.org/10.3390/ma12081253
Kiener J, Limousy L, Jeguirim M, Le Meins J-M, Hajjar-Garreau S, Bigoin G, Ghimbeu CM. Activated Carbon/Transition Metal (Ni, In, Cu) Hexacyanoferrate Nanocomposites for Cesium Adsorption. Materials. 2019; 12(8):1253. https://doi.org/10.3390/ma12081253
Chicago/Turabian StyleKiener, Julien, Lionel Limousy, Mejdi Jeguirim, Jean-Marc Le Meins, Samar Hajjar-Garreau, Gaetan Bigoin, and Camélia Matei Ghimbeu. 2019. "Activated Carbon/Transition Metal (Ni, In, Cu) Hexacyanoferrate Nanocomposites for Cesium Adsorption" Materials 12, no. 8: 1253. https://doi.org/10.3390/ma12081253
APA StyleKiener, J., Limousy, L., Jeguirim, M., Le Meins, J.-M., Hajjar-Garreau, S., Bigoin, G., & Ghimbeu, C. M. (2019). Activated Carbon/Transition Metal (Ni, In, Cu) Hexacyanoferrate Nanocomposites for Cesium Adsorption. Materials, 12(8), 1253. https://doi.org/10.3390/ma12081253