Correlation of Morphology and In-Vitro Degradation Behavior of Spray Pyrolyzed Bioactive Glasses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Characterization
3. Results
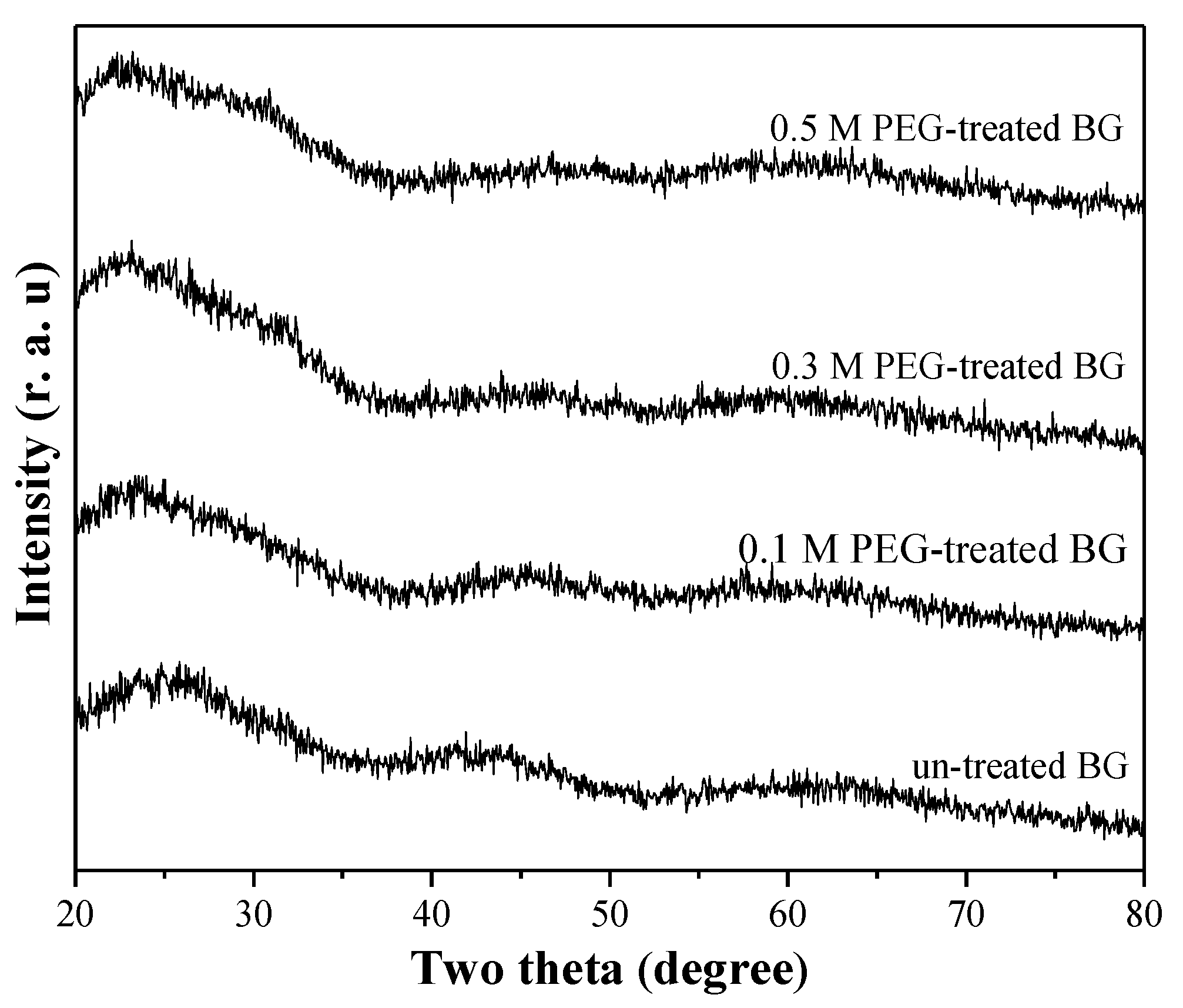
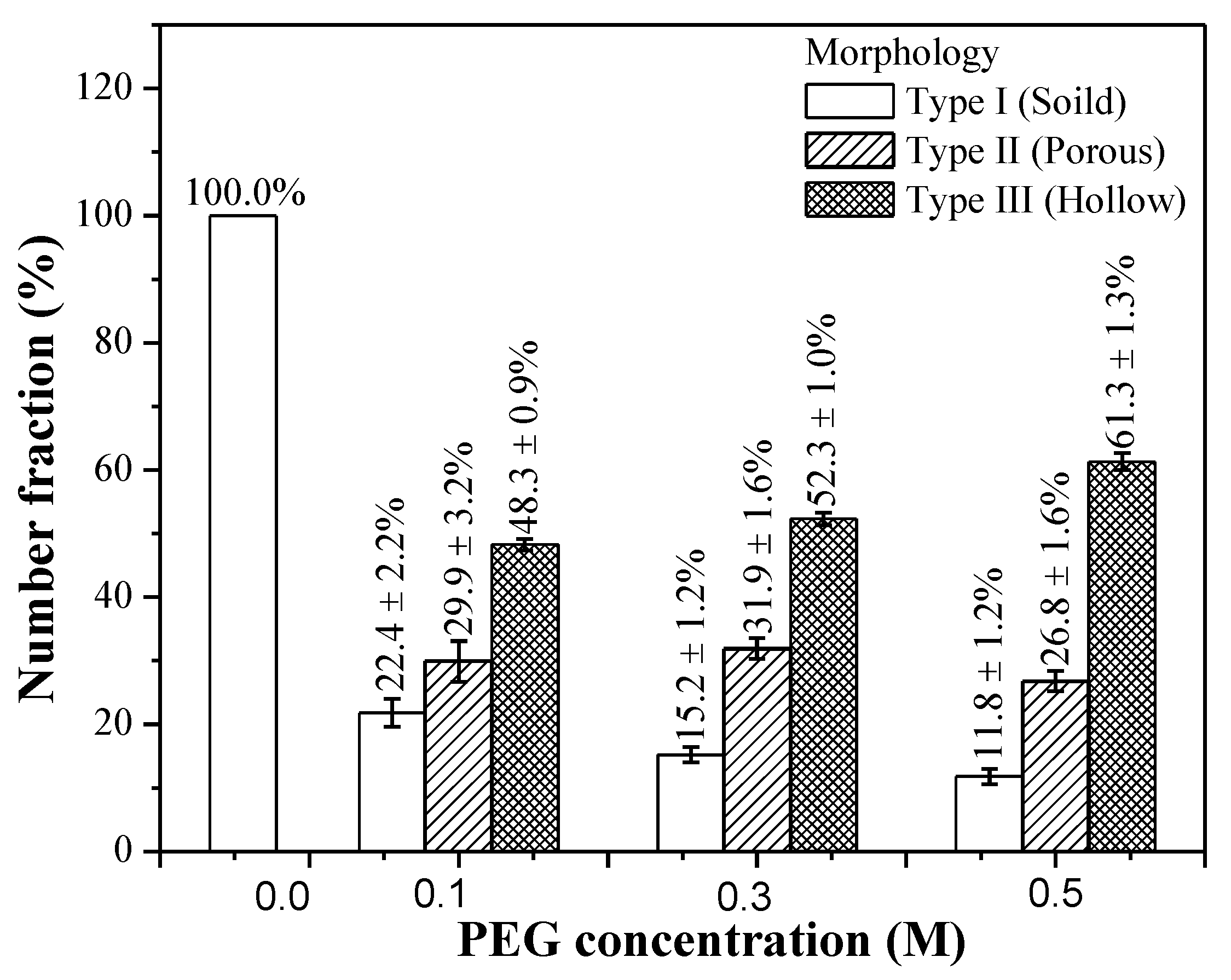
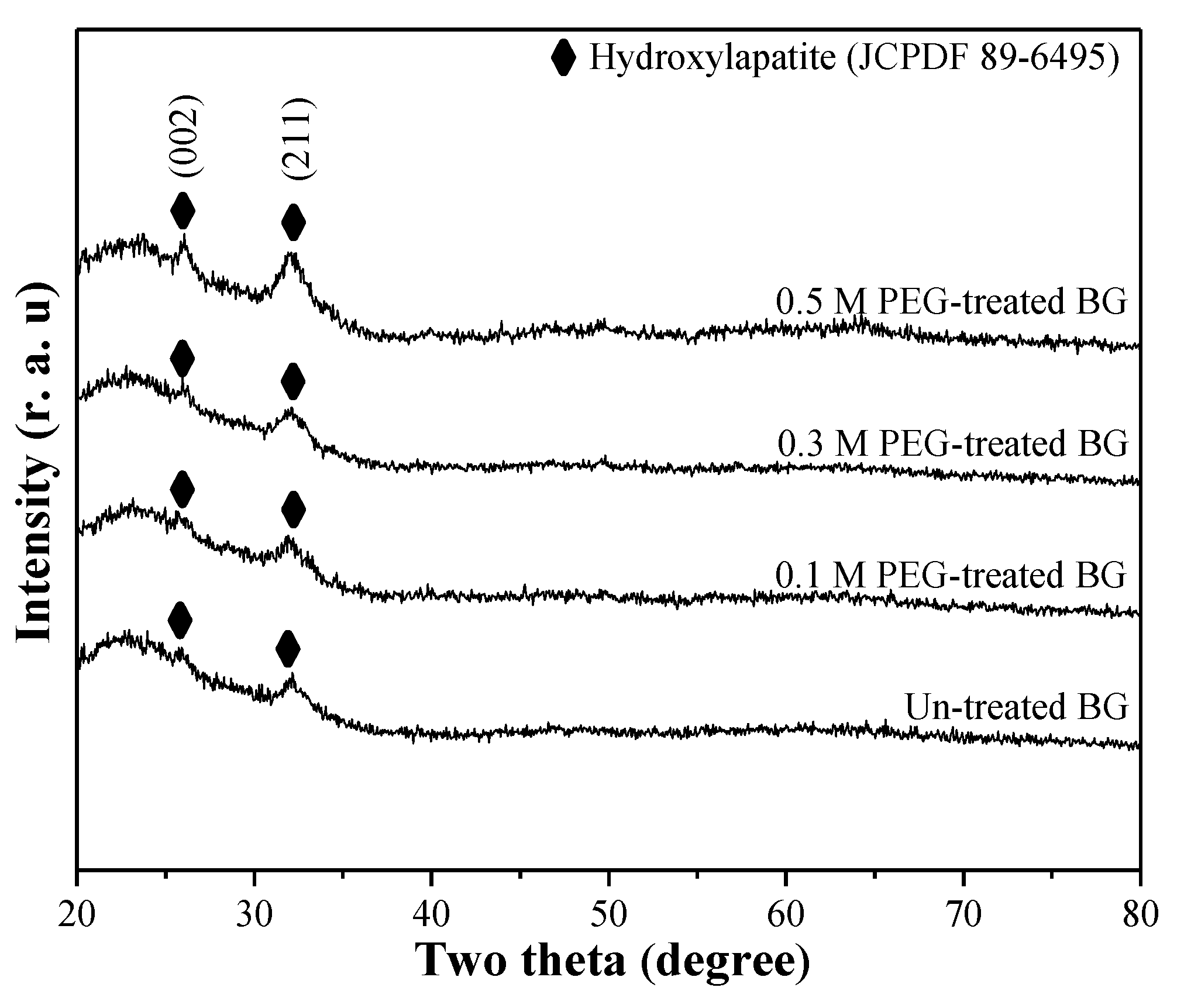
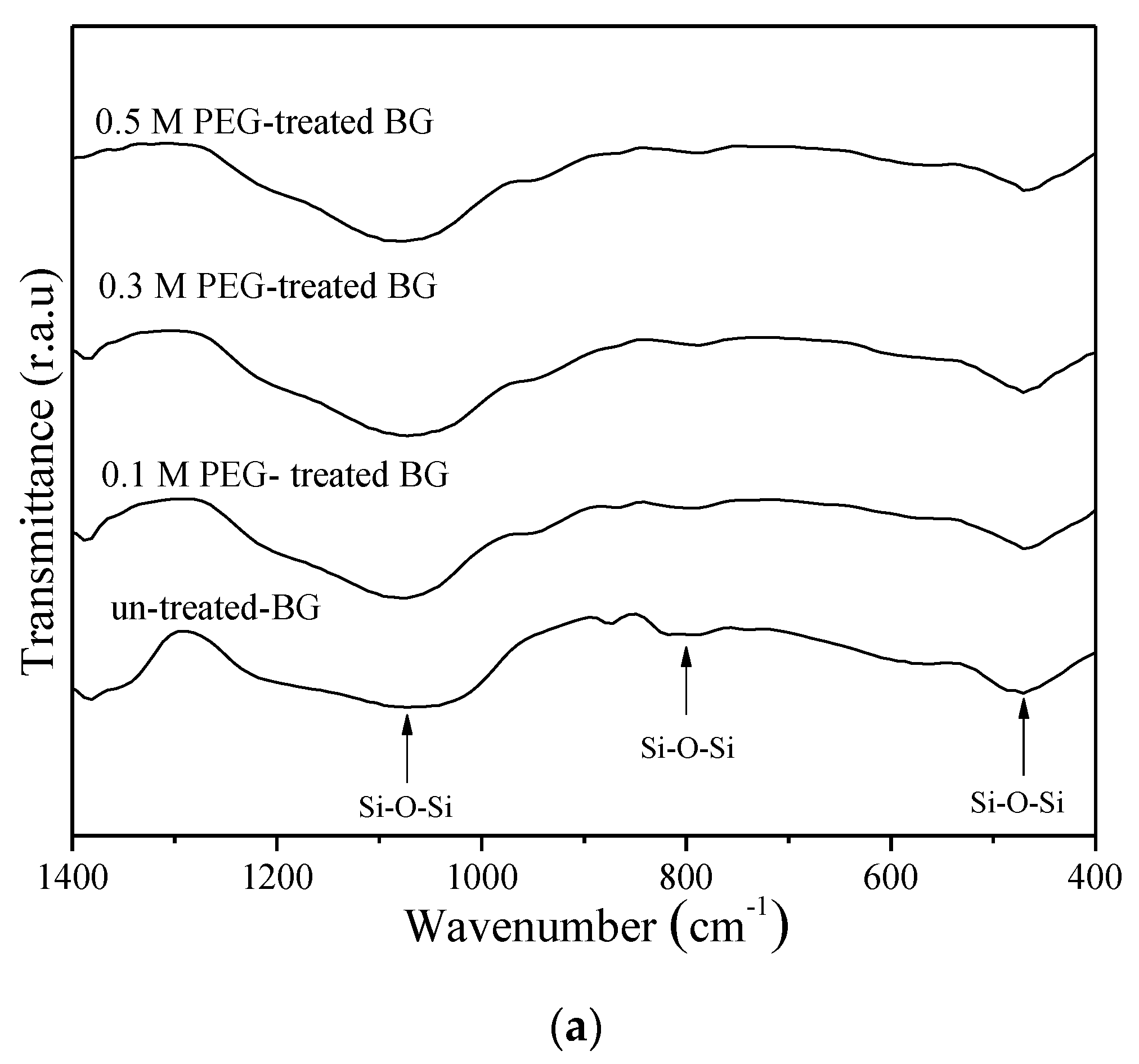
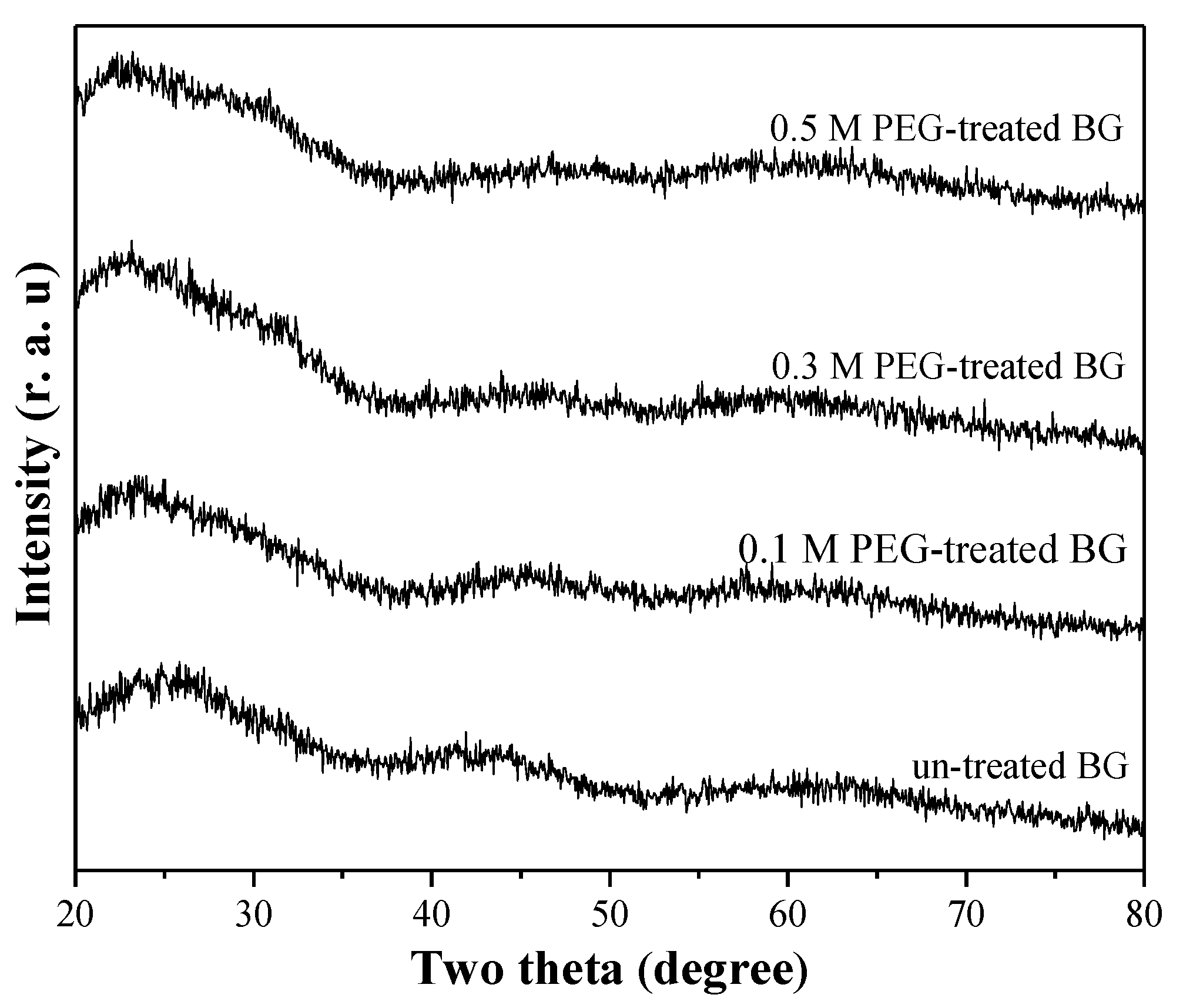
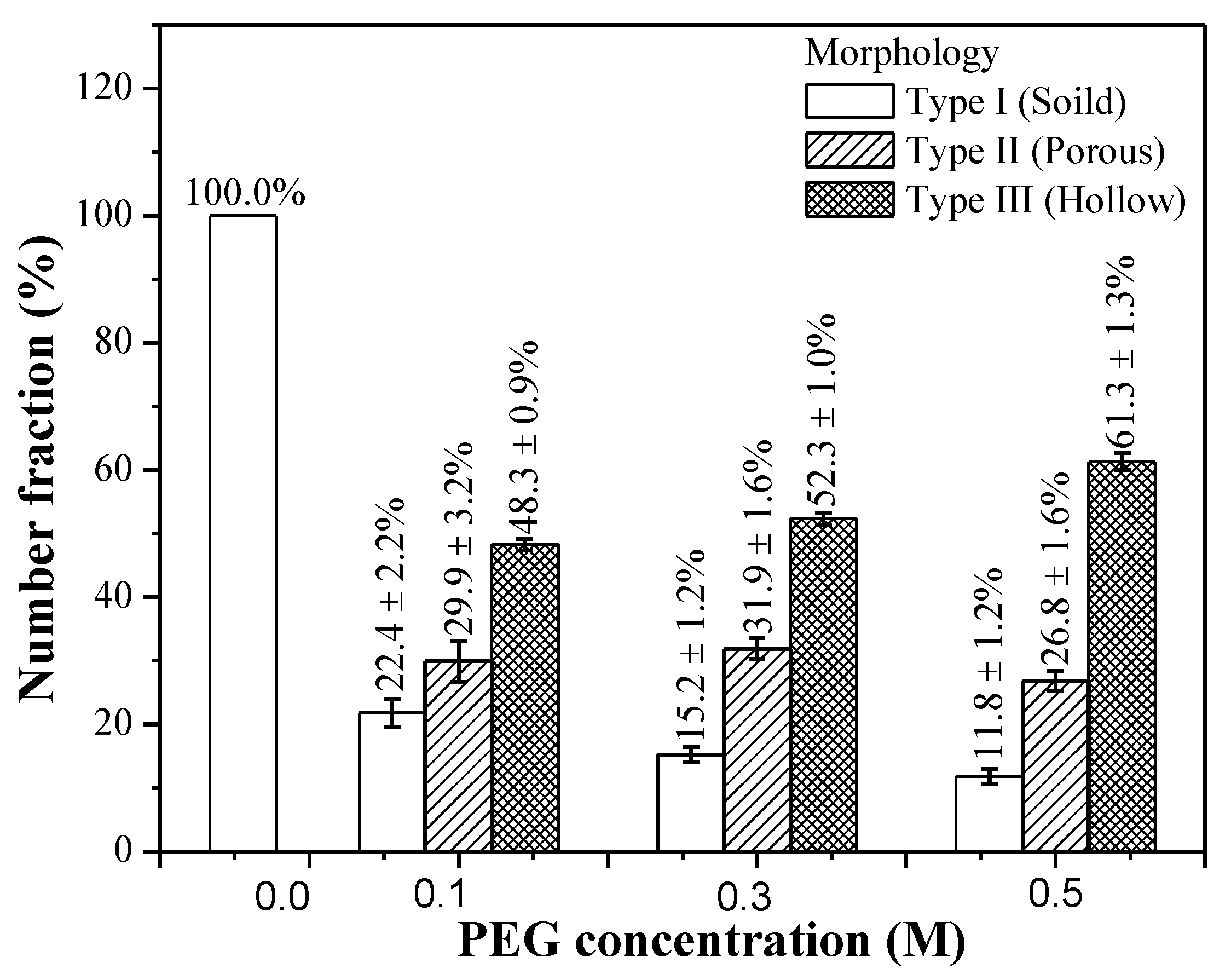
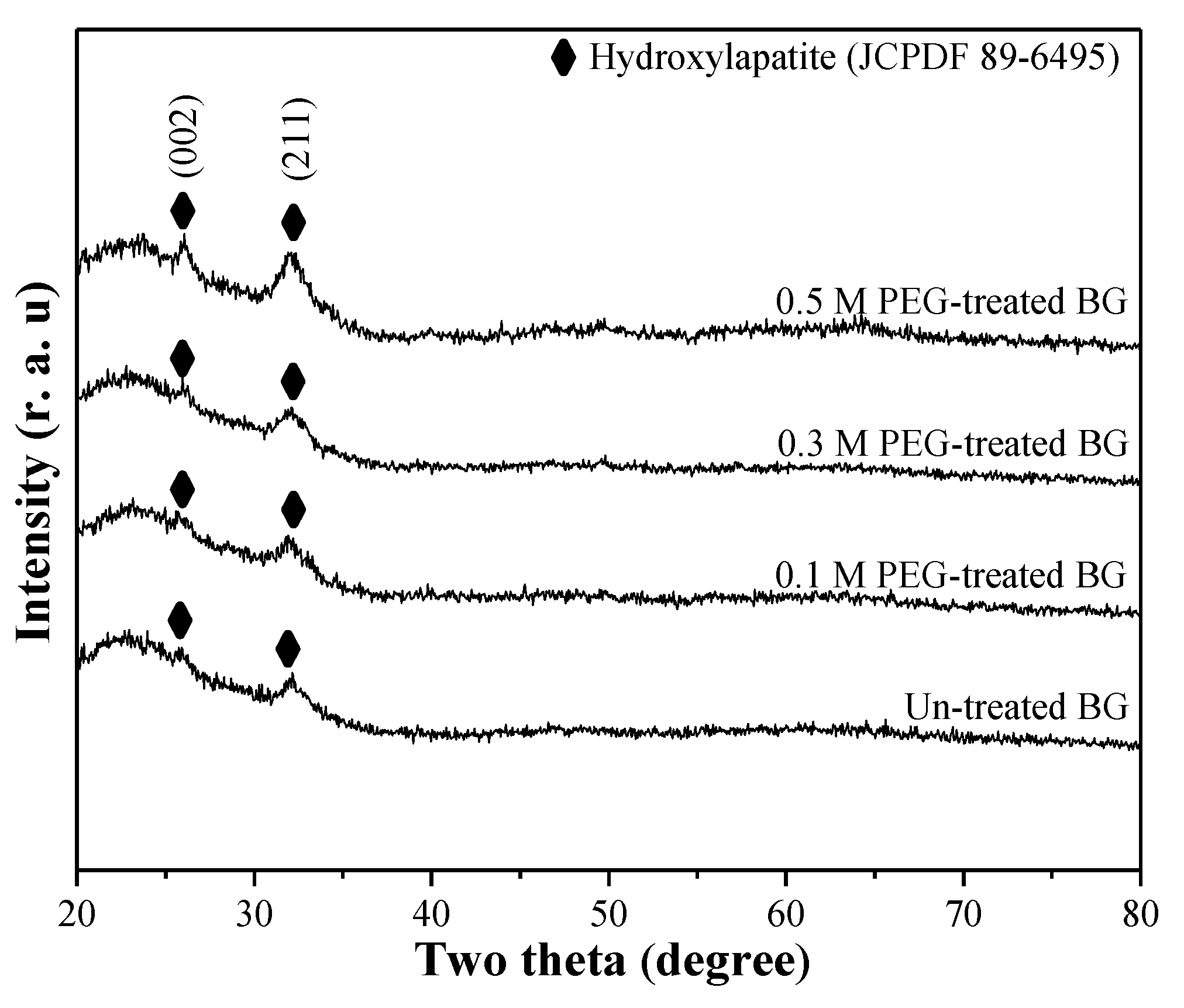
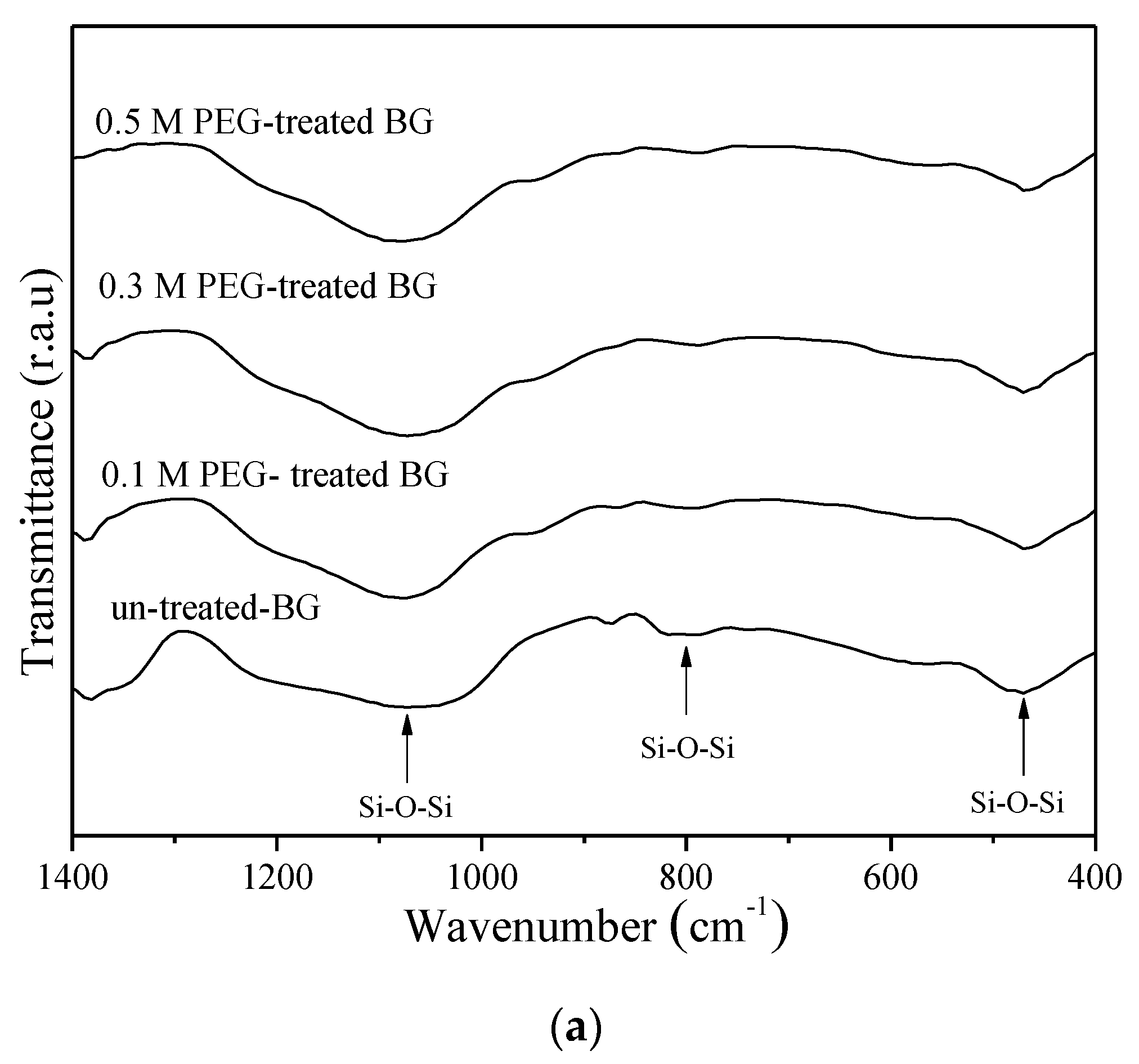
3.1. Phase Composition and Morphology
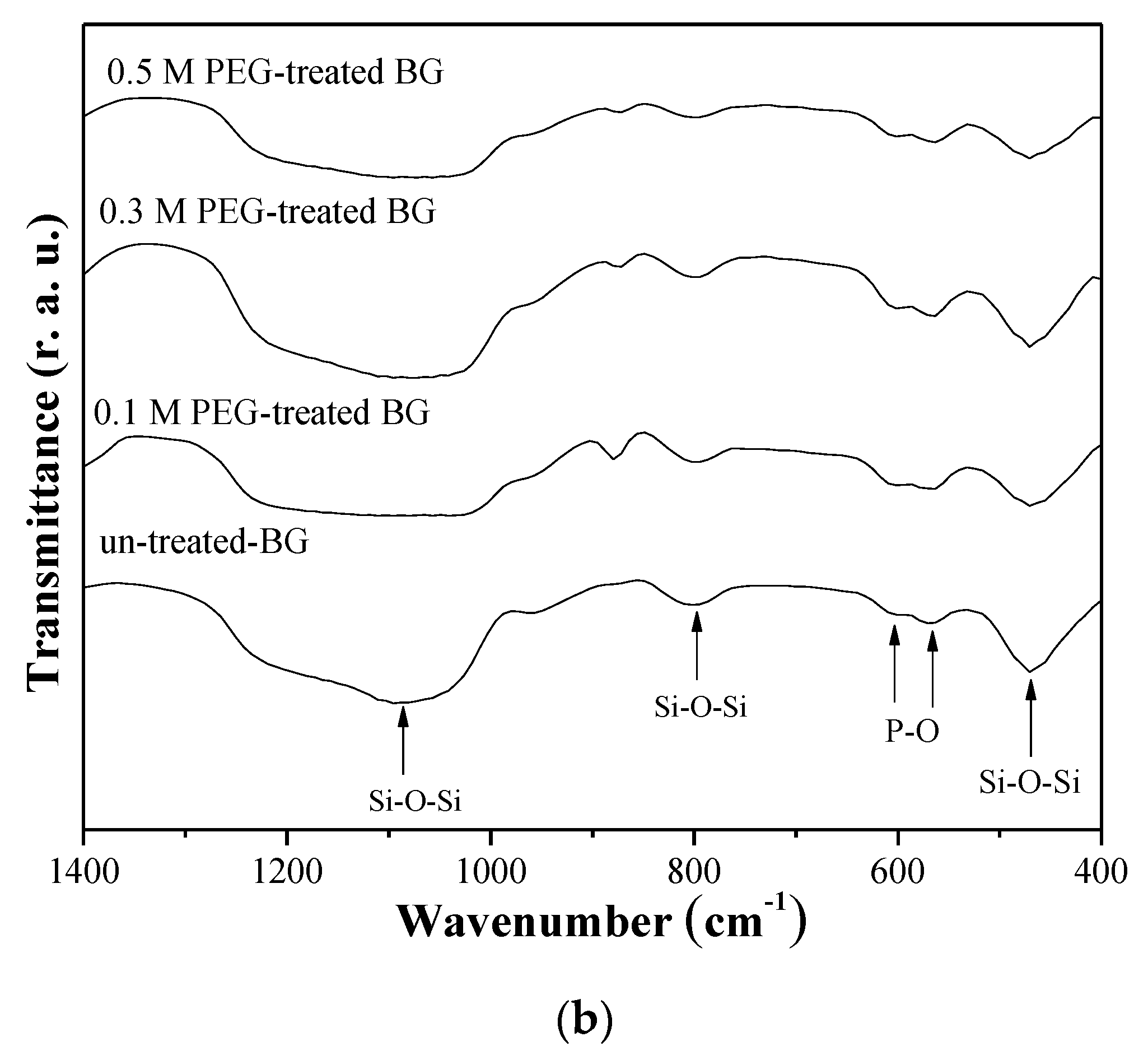
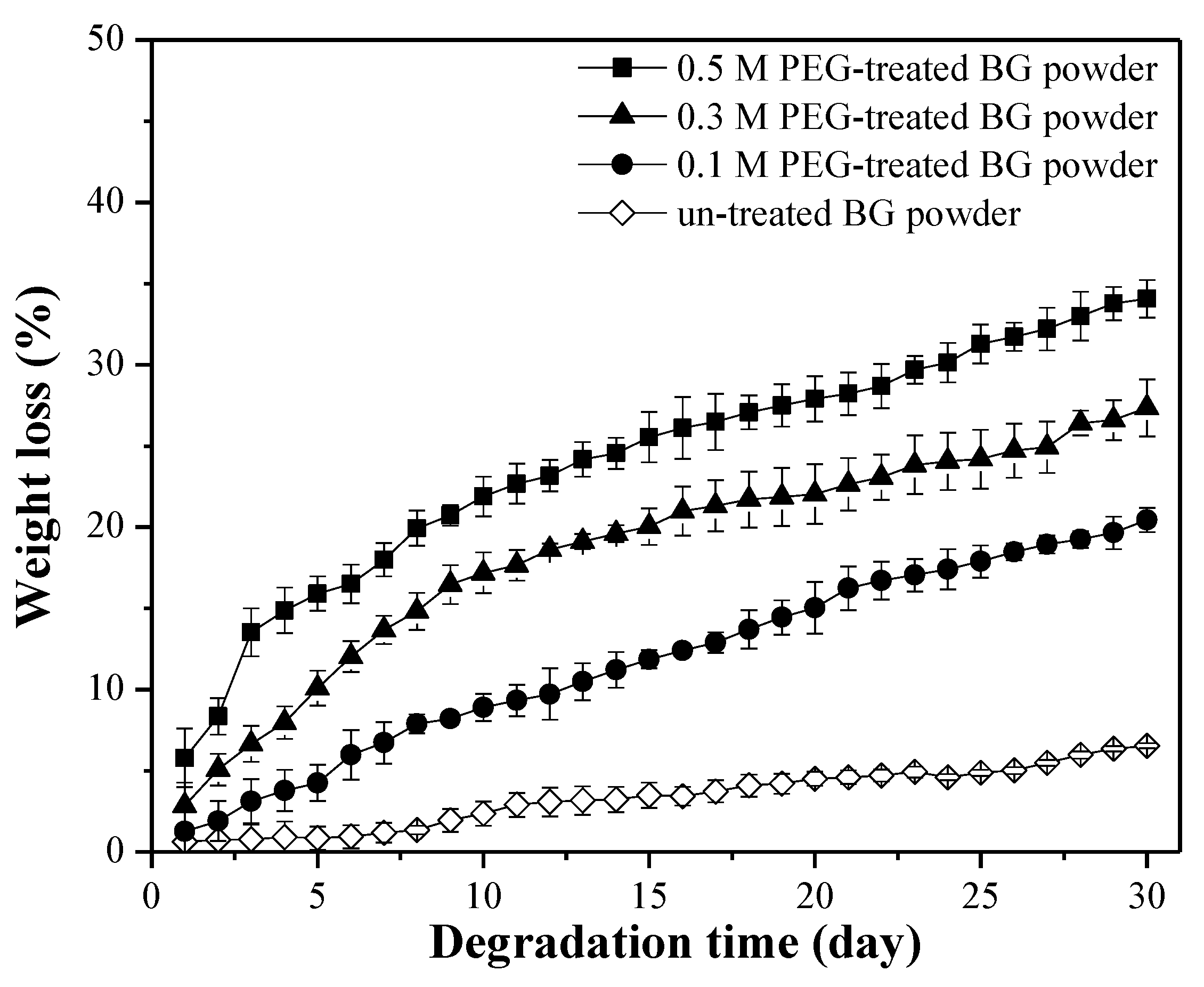
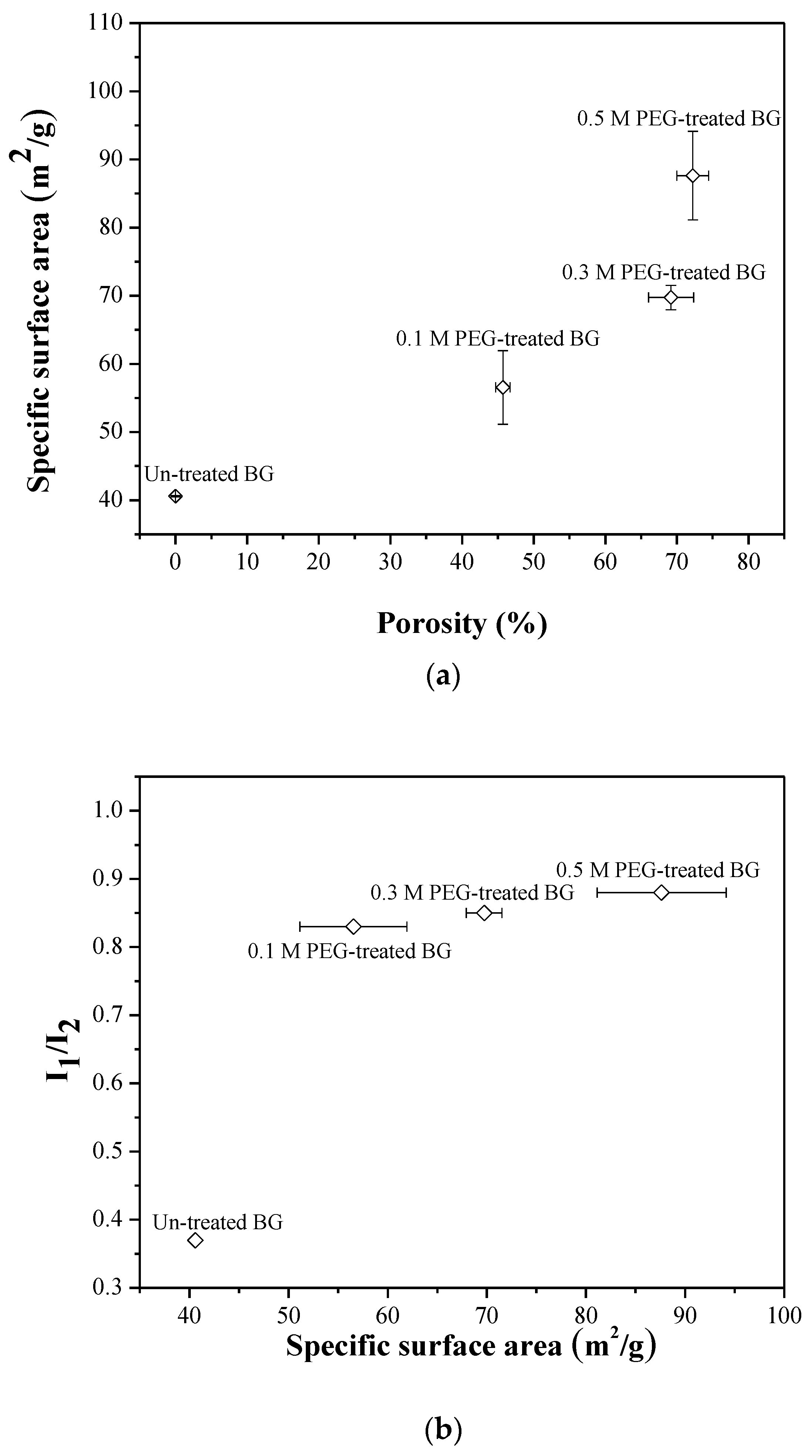
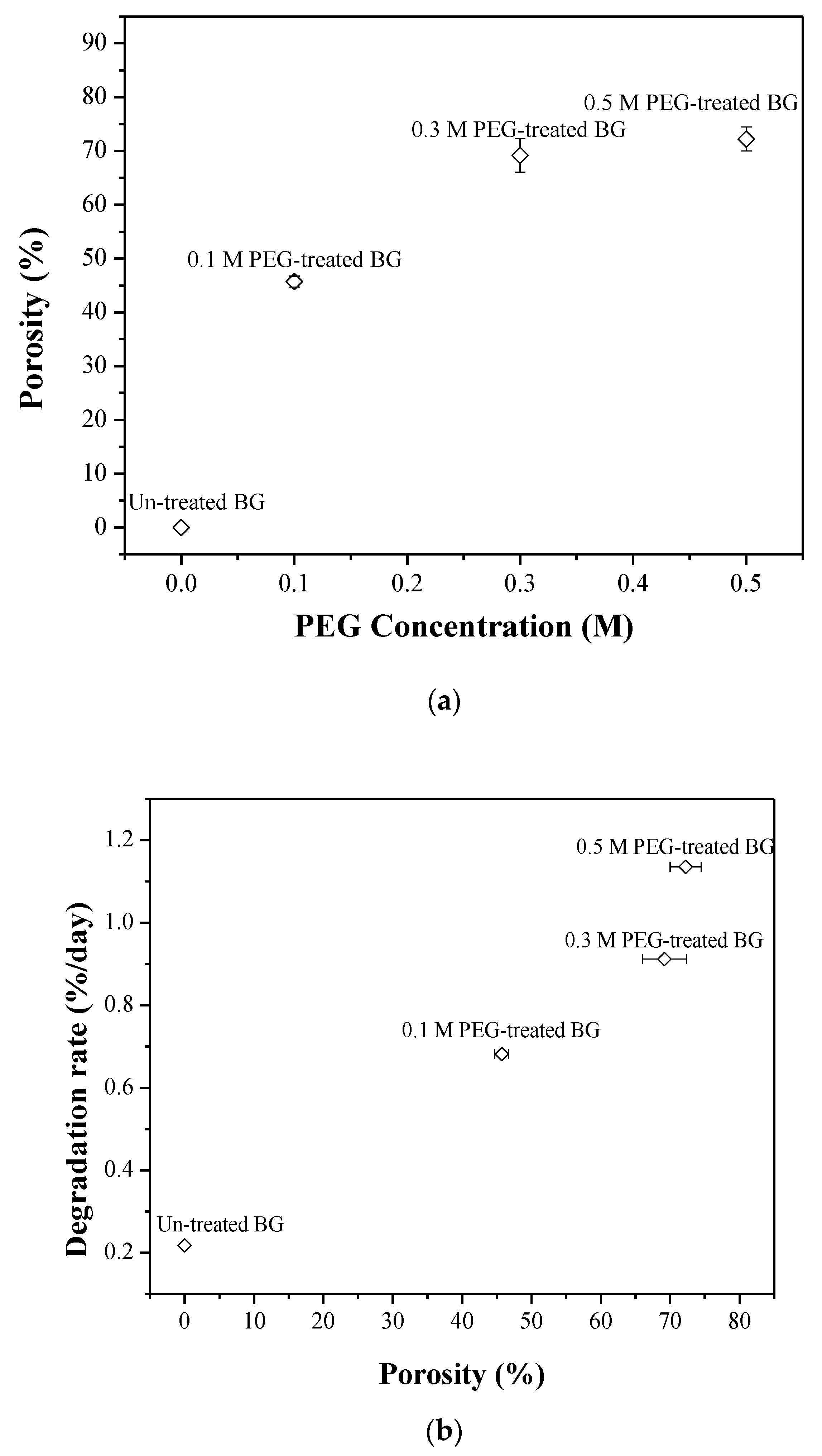
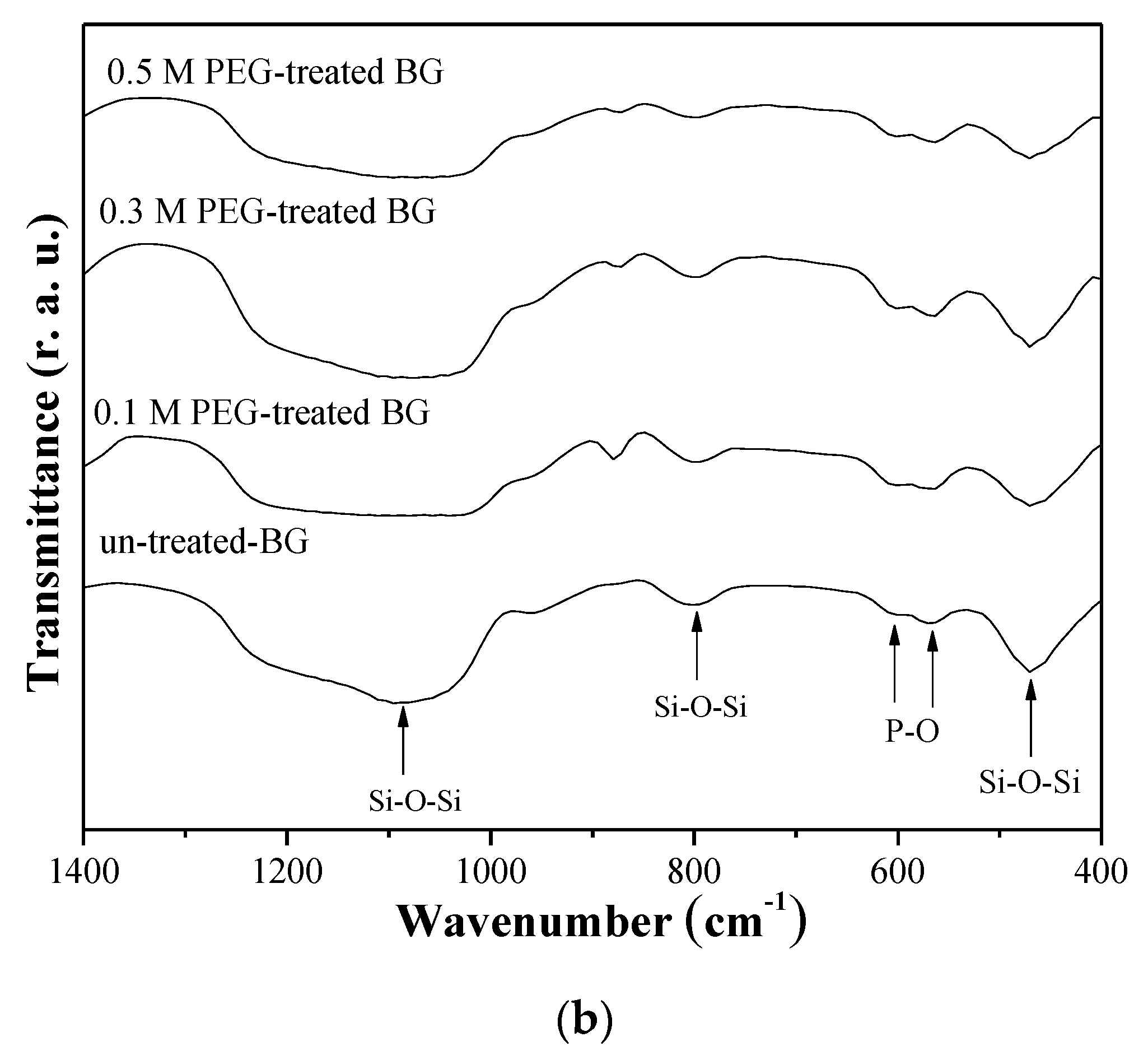
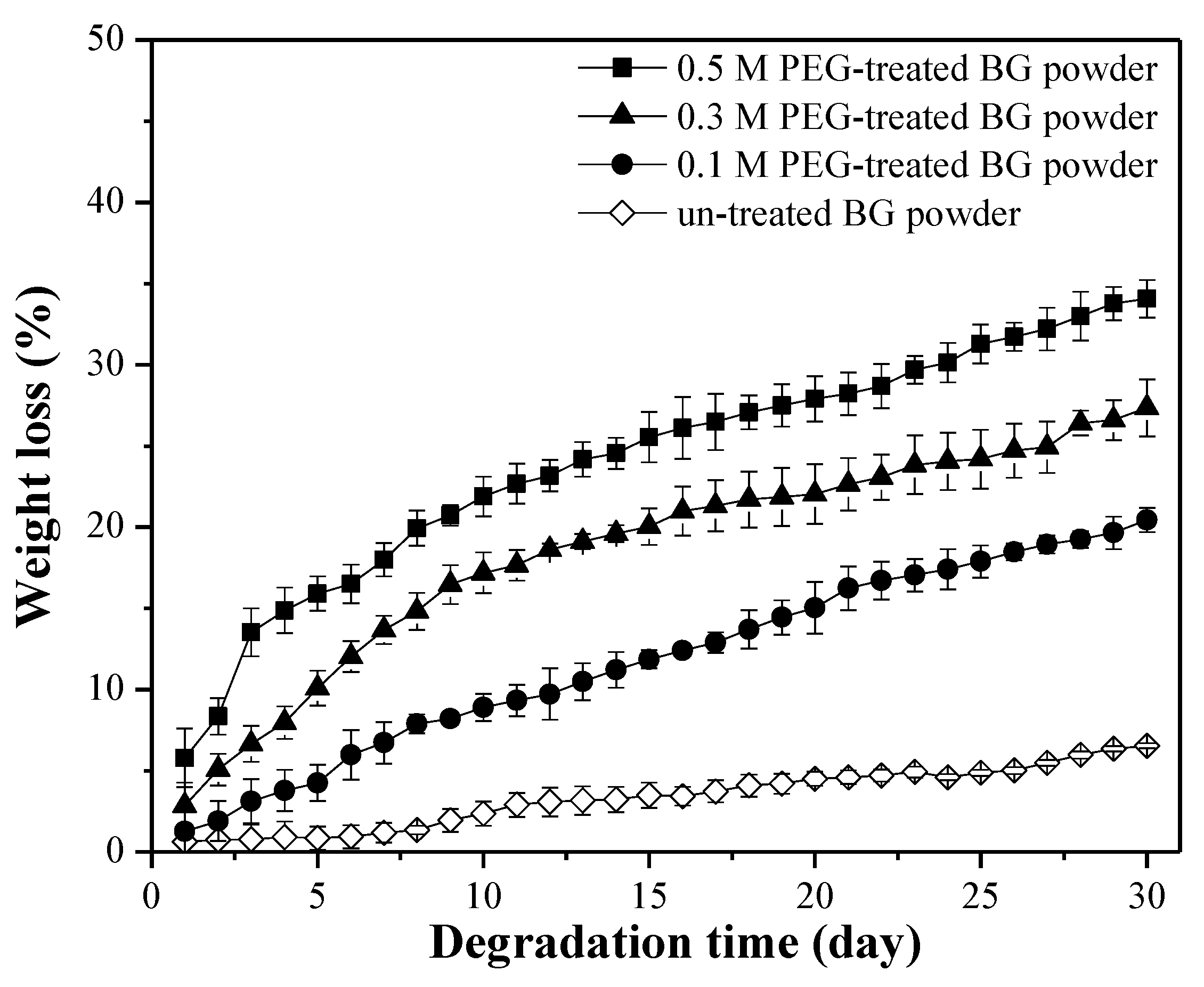
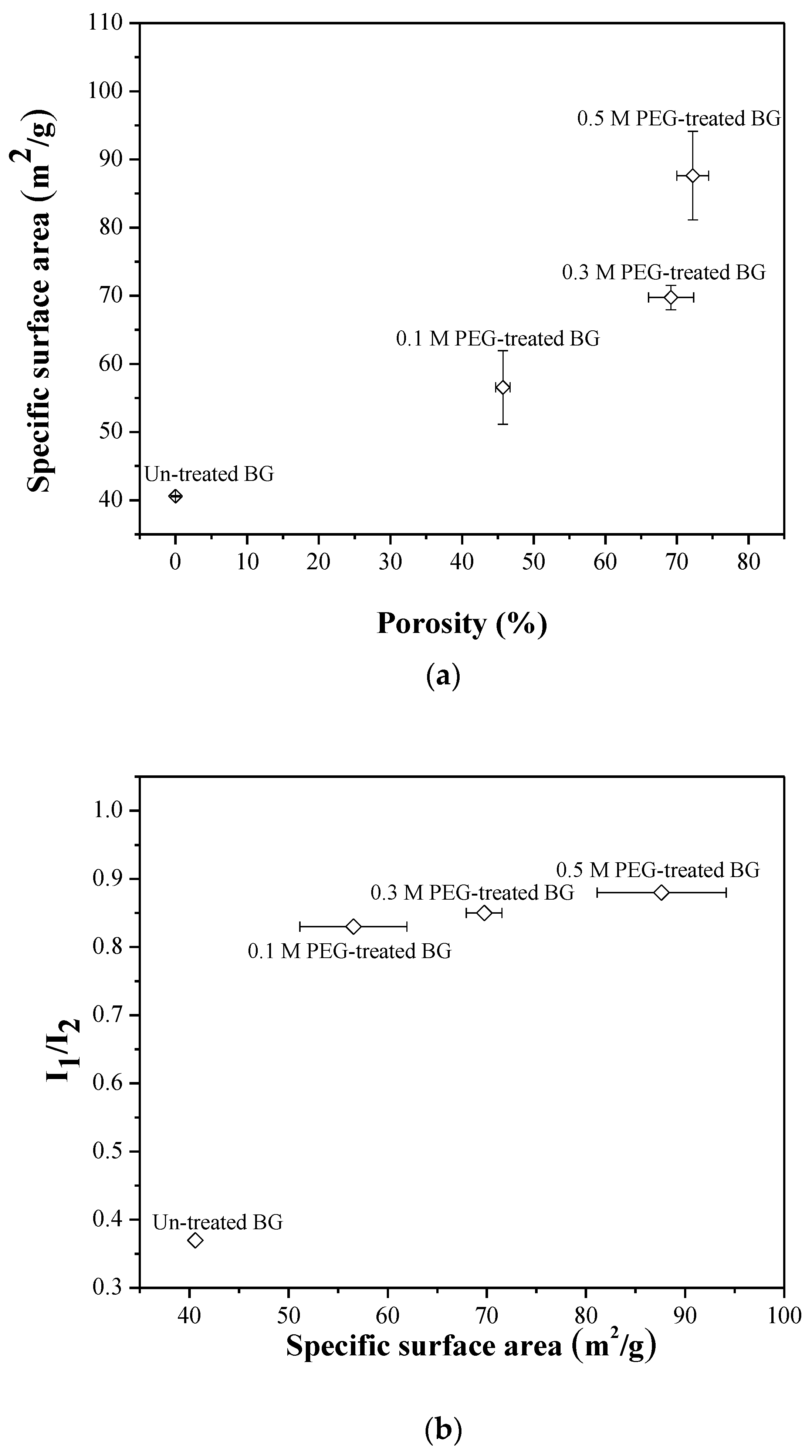
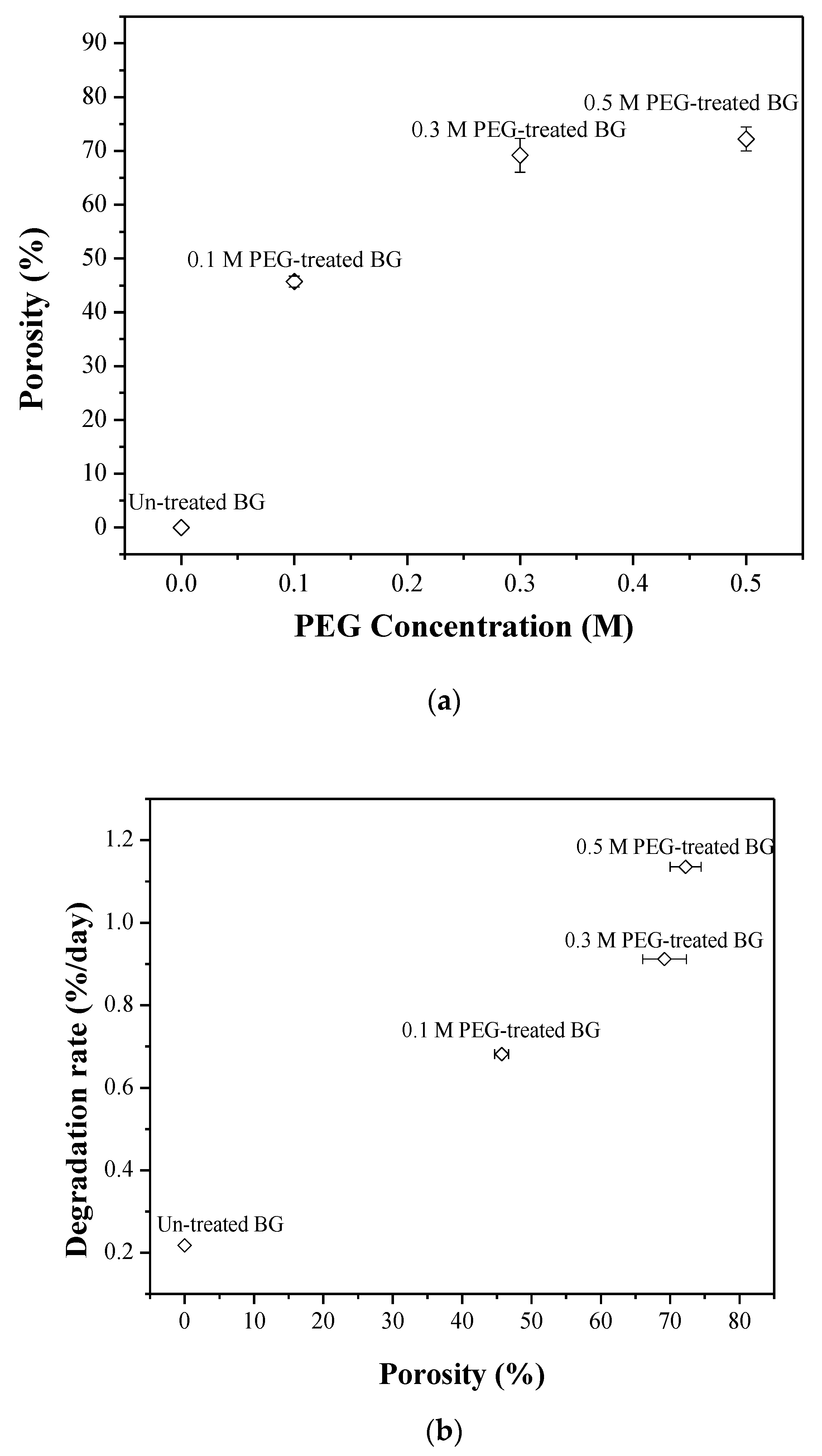
3.2. Specific Surface Area and Bioactivity, In-Vitro Degradation Test
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Catteaux, R.; Lebecq, I.; Désanglois, F.; Chai, F.; Hornez, J.-C.; Hampshire, S.; Follet, C. Synthesis, characterization and bioactivity of bioglasses in the na2o–cao–p2o5–sio2 system prepared via sol gel processing. Chem. Eng. Res. Des. 2013, 91, 2420–2426. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics: From concept to clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Bellucci, D.; Salvatori, R.; Giannatiempo, J.; Anesi, A.; Bortolini, S.; Cannillo, V. A new bioactive glass/collagen hybrid composite for applications in dentistry. Materials 2019, 12, 2079. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Chen, X.; Wang, Y.; Zhao, N.; Miao, G.; Li, Z.; Lin, C. Fabrication of porous bioactive glass particles by one step sintering. Mater. Lett. 2010, 64, 2293–2295. [Google Scholar] [CrossRef]
- Lei, B.; Chen, X.; Wang, Y.; Zhao, N.; Du, C.; Fang, L. Surface nanoscale patterning of bioactive glass to support cellular growth and differentiation. J. Biomed. Mater Res. Part A 2010, 94, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Fiume, E.; Barberi, J.; Verné, E.; Baino, F. Bioactive glasses: From parent 45S5 composition to scaffold-assisted tissue-healing therapies. J. Funct. Biomater. 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Fujibayashi, S.; Neo, M.; Kim, H.-M.; Kokubo, T.; Nakamura, T. A comparative study between in vivo bone ingrowth and in vitro apatite formation on Na2O–CaO–SiO2 glasses. Biomaterials 2003, 24, 1349–1356. [Google Scholar] [CrossRef]
- Hill, R. An alternative view of the degradation of bioglass. J. Mater. Sci. Lett. 1996, 15, 1122–1125. [Google Scholar] [CrossRef]
- Zhang, D.; Jain, H.; Hupa, M.; Hupa, L.; Bandyopadhyay, A. In-vitro degradation and bioactivity of tailored amorphous multi porous scaffold structure. J. Am. Chem. Soc. 2012, 95, 2687–2694. [Google Scholar] [CrossRef]
- Deliormanlı, A.M. Size-dependent degradation and bioactivity of borate bioactive glass. Ceram. Int. 2013, 39, 8087–8095. [Google Scholar] [CrossRef]
- Hench, L.L.; West, J.K. The sol-gel process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Lei, B.; Chen, X.; Wang, Y.; Zhao, N.; Du, C.; Fang, L. Fabrication, structure and biological properties of organic acid-derived sol-gel bioactive glasses. Biomed. Mater. 2010, 5, 054103. [Google Scholar] [CrossRef] [PubMed]
- Sales, M.; Alarcon, J. Crystallization of sol-gel derived glass ceramic powders in the CaO-MgO-Al2O3-SiO2 system. J. Mater. Sci. 1995, 30, 2341–2347. [Google Scholar] [CrossRef]
- Molino, G.; Bari, A.; Baino, F.; Fiorilli, S.; Vitale-Brovarone, C. Electrophoretic deposition of spray-dried sr-containing mesoporous bioactive glass spheres on glass–ceramic scaffolds for bone tissue regeneration. J. Mater. Sci. 2017, 52, 9103–9114. [Google Scholar] [CrossRef]
- Chou, Y.-J.; Hsiao, C.-W.; Tsou, N.-T.; Wu, M.-H.; Shih, S.-J. Preparation and in vitro bioactivity of micron-sized bioactive glass particles using spray drying method. Appl. Sci. 2018, 9, 19. [Google Scholar] [CrossRef]
- Hong, B.-J.; Shih, S.-J. Novel pore-forming agent to prepare of mesoporous bioactive glass using one-step spray pyrolysis. Ceram. Int. 2017, 43, S771–S775. [Google Scholar] [CrossRef]
- Shih, S.-J.; Sari, D.R.M.; Lin, Y.C. Influence of chemical composition on the bioactivity of spray pyrolyzed mesoporous bioactive glass. Int. J. Appl. Ceram. Technol. 2016, 13, 787–794. [Google Scholar] [CrossRef]
- Lei, B.; Chen, X.; Wang, Y.; Zhao, N. Synthesis and in vitro bioactivity of novel mesoporous hollow bioactive glass microspheres. Mater. Lett. 2009, 63, 1719–1721. [Google Scholar] [CrossRef]
- Lei, B.; Shin, K.H.; Moon, Y.W.; Noh, D.Y.; Koh, Y.H.; Jin, Y.; Kim, H.E.; Ducheyne, P. Synthesis and bioactivity of sol–gel derived porous, bioactive glass microspheres using chitosan as novel biomolecular template. J. Am. Ceram. Soc. 2012, 95, 30–33. [Google Scholar] [CrossRef]
- Sharma, M.K.; Rohani, P.; Liu, S.; Kaus, M.; Swihart, M.T. Polymer and surfactant-templated synthesis of hollow and porous zns nano- and microspheres in a spray pyrolysis reactor. Langmuir 2015, 31, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, A.H.; Chou, Y.-J.; Tan, C.-Y.; Bakare, F.F.; Tsou, N.-T.; Huang, E.W.; Shih, S.-J. Control of dopant distribution in yttrium-doped bioactive glass for selective internal radiotherapy applications using spray pyrolysis. Materials 2019, 12, 986. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, C.Q.; Pederson, L.R.; McVay, G.L. The effects of surface area to solution volume ratio and surface roughness on glass leaching. J. Non-Cryst. Solids 1982, 49, 397–412. [Google Scholar] [CrossRef]
- Shih, S.-J.; Herrero, P.R.; Li, G.; Chen, C.-Y.; Lozano-Perez, S. Characterization of mesoporosity in ceria particles using electron microscopy. Microsc. Microanal. 2010, 17, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Messing, G.L.; Zhang, S.-C.; Jayanthi, G.V. Ceramic powder synthesis by spray pyrolysis. J. Am. Ceram. Soc. 1993, 76, 2707–2726. [Google Scholar] [CrossRef]
- Kokubo, T.; Kushitani, H.; Ohtsuki, C.; Sakka, S.; Yamamuro, T. Chemical reaction of bioactive glass and glass-ceramics with a simulated body fluid. J. Mater. Sci. Mater. Med. 1992, 3, 79–83. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using castep. Z. Kristallogr. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Arenas, L.T.; Simm, C.W.; Gushikem, Y.; Dias, S.L.P.; Moro, C.C.; Costa, T.M.H.; Benvenutti, E.V. Synthesis of silica xerogels with high surface area using acetic acid as catalyst. J. Braz. Chem. Soc. 2007, 18, 886–890. [Google Scholar] [CrossRef]
- King, P.L.; Ramsey, M.S.; Swayze, G.A. Infrared Spectroscopy in Geochemistry, Exploration Geochemistry and Remote Sensing; Mineralogical Association of Canada: Quebec, QC, Canada, 2004; Volume 33. [Google Scholar]
- King, P.; McMillan, P.; Moore, G.; Ramsey, M.; Swayze, G. Infrared Spectroscopy of Silicate Glasses with Application to Natural Systems; Mineralogical Association of Canada: Quebec, QC, Canada, 2004; pp. 93–133. [Google Scholar]
- Shih, S.-J.; Chou, Y.-J.; Chen, C.-Y.; Lin, C.-K. One-step synthesis and characterization of nanosized bioactive glass. J. Med. Biol. Eng. 2014, 34, 18–23. [Google Scholar] [CrossRef]
- Shih, S.-J.; Chou, Y.-J.; Hadush, A.; Lin, S.-H.; Hsiao, C.-W. Morphology control of eu-doped amorphous gehlenite phosphors prepared by spray pyrolysis. J. Nanosci. Nanotechnol. 2018, 18, 5849–5853. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.-J.; Chou, Y.-J.; Borisenko, K.B. Preparation method: Structure–bioactivity correlation in mesoporous bioactive glass. J. Nanopart. Res. 2013, 15, 1763. [Google Scholar] [CrossRef]
- Qi, X.; Pei, P.; Zhu, M.; Du, X.; Xin, C.; Zhao, S.; Li, X.; Zhu, Y. Three dimensional printing of calcium sulfate and mesoporous bioactive glass scaffolds for improving bone regeneration in vitro and in vivo. Sci. Rep. 2017, 7, 42556. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakare, F.F.; Chou, Y.-J.; Huang, Y.-H.; Tesfay, A.H.; Moriga, T.; Shih, S.-J. Correlation of Morphology and In-Vitro Degradation Behavior of Spray Pyrolyzed Bioactive Glasses. Materials 2019, 12, 3703. https://doi.org/10.3390/ma12223703
Bakare FF, Chou Y-J, Huang Y-H, Tesfay AH, Moriga T, Shih S-J. Correlation of Morphology and In-Vitro Degradation Behavior of Spray Pyrolyzed Bioactive Glasses. Materials. 2019; 12(22):3703. https://doi.org/10.3390/ma12223703
Chicago/Turabian StyleBakare, Fetene Fufa, Yu-Jen Chou, Yu-Hsuan Huang, Abadi Hadush Tesfay, Toshihiro Moriga, and Shao-Ju Shih. 2019. "Correlation of Morphology and In-Vitro Degradation Behavior of Spray Pyrolyzed Bioactive Glasses" Materials 12, no. 22: 3703. https://doi.org/10.3390/ma12223703
APA StyleBakare, F. F., Chou, Y.-J., Huang, Y.-H., Tesfay, A. H., Moriga, T., & Shih, S.-J. (2019). Correlation of Morphology and In-Vitro Degradation Behavior of Spray Pyrolyzed Bioactive Glasses. Materials, 12(22), 3703. https://doi.org/10.3390/ma12223703