Shrinkage and Mitigation Strategies to Improve the Dimensional Stability of CaO-FeOx-Al2O3-SiO2 Inorganic Polymers

 ,
, 
Abstract
1. Introduction
2. Experiments
2.1. Materials
2.2. Methods
2.3. Experimental Conditions
3. Results and Discussion
3.1. Precursors’ Characterization
3.2. Isothermal Conduction Calorimetry Testing (ICC)
3.3. XRD Analysis
3.4. ATR-FTIR Analysis
3.5. Shrinkage Behavior of IPs Mortars
3.5.1. Autogenous Shrinkage
3.5.2. Drying Shrinkage and Weight Loss
3.6. Mercury Intrusion Porosimetry
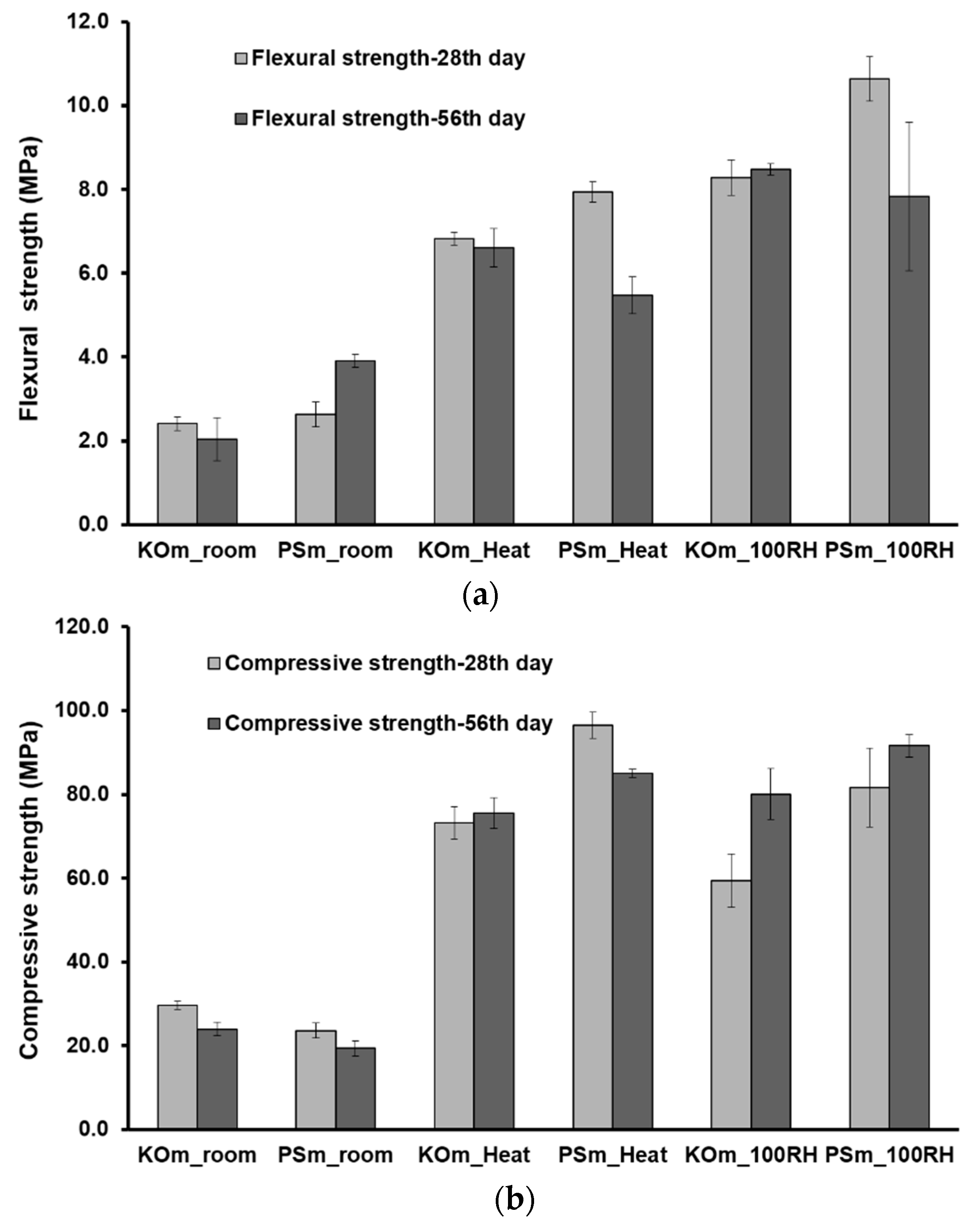
3.7. Mechanical Properties
4. Conclusions
- The precursors’ reactivity and curing conditions severely affected shrinkage mechanisms and its magnitude in IP mortars.
- The volumetric changes that occurred during the binders’ plastic stage were defined by the precursors’ reactivity. In the case studies analyzed here, considerable autogenous shrinkage or expansion has been observed in IP mortars.
- At room condition in a hardened stage, regardless of the precursors’ bulk composition, drying shrinkage was identified as the governing mechanism affecting the mortars’ volumetric stability, whereas autogenous shrinkage was less significant.
- The characteristics of the precursors affected the reaction kinetics and porous structures formed, thus modifying the mortars’ pore size distribution and greatly influencing their drying shrinkage behavior.
- Thermal treatment promoted a decrease in porosity and the redistribution of pores to lower dimensions, but it was found to be an effective shrinkage-control strategy as it reduced the total shrinkage of IP mortars by 30%. As expected, in water-saturated curing conditions, drying shrinkage was found to be negligible and corresponded to the results of corrugated tube autogenous shrinkage in the hardened stage.
- Thermal and moist curing promoted higher volumetric stability and considerably improved themechanical features of IP mortars. Mortars with enhanced flexural (up to 10.7 MPa) and compressive strength (up to 81.7 MPa) were produced.
Author Contributions
Funding
Conflicts of Interest
References
- Xu, G.; Shi, X. Characteristics and applications of fly ash as a sustainable construction material: A state-of-the-art review. Resour. Conserv. Recycl. 2018, 136, 95–109. [Google Scholar] [CrossRef]
- Golewski, G.L. Effect of fly ash addition on the fracture toughness of plain concrete at third model of fracture. J. Civ. Eng. Manag. 2017, 23, 613–620. [Google Scholar] [CrossRef]
- Ascensão, G.; Marchi, M.; Segata, M.; Faleschini, F.; Pontikes, Y. Reaction kinetics and structural analysis of alkali activated Fe-Si-Ca rich materials. J. Clean. Prod. 2019, in press. [Google Scholar]
- Provis, J.L. Alkali-activated materials. Cement Concr. Res. 2018, 114, 40–48. [Google Scholar] [CrossRef]
- Mastali, M.; Kinnunen, P.; Dalvand, A.; Firouz, R.M.; Illikainen, M. Drying shrinkage in alkali-activated binders—A critical review. Constr. Build. Mater. 2018, 190, 533–550. [Google Scholar] [CrossRef]
- Hertel, T.; Novais, R.M.; Alarcón, R.M.; Labrincha, J.A.; Pontikes, Y. Use of modified bauxite residue-based porous inorganic polymer monoliths as adsorbents of methylene blue. J. Clean. Prod. 2019, 227, 877–889. [Google Scholar] [CrossRef]
- Ascensão, G.; Seabra, M.P.; Aguiar, J.B.; Labrincha, J.A. Red mud-based geopolymers with tailored alkali diffusion properties and pH buffering ability. J. Clean. Prod. 2017, 148, 23–30. [Google Scholar] [CrossRef]
- Okada, K.; Isobe, T.; Katsumata, K.I.; Kameshima, Y.; Nakajima, A.; MacKenzie, K.J. Porous ceramics mimicking nature—Preparation and properties of microstructures with unidirectionally oriented pores. Sci. Technol. Adv. Mater. 2011, 12, 064701. [Google Scholar] [CrossRef]
- Ge, Y.; Yuan, Y.; Wang, K.; He, Y.; Cui, X. Preparation of geopolymer-based inorganic membrane for removing Ni2+ from wastewater. J. Hazard Mater. 2015, 299, 711–718. [Google Scholar] [CrossRef]
- Papa, E.; Medri, V.; Benito, P.; Vaccari, A.; Bugani, S.; Jaroszewicz, J.; Landi, E. Synthesis of porous hierarchical geopolymer monoliths by ice-templating. Microporous Mesoporous Mater. 2015, 215, 206–214. [Google Scholar] [CrossRef]
- Zhang, Z.; Provis, J.L.; Reid, A.; Wang, H. Mechanical, thermal insulation, thermal resistance and acoustic absorption properties of geopolymer foam concrete. Cement Concr. Comp. 2015, 62, 97–105. [Google Scholar] [CrossRef]
- Mast, B.; Gerardy, I.; Pontikes, Y.; Schroeyers, W.; Reniers, B.; Samyn, P.; Schreurs, S. The effect of gamma radiation on the mechanical and microstructural properties of Fe-rich inorganic polymers. J. Nucl. Mater. 2019, 521, 126–136. [Google Scholar] [CrossRef]
- Naser, M.Z. Extraterrestrial construction materials. Prog. Mater. Sci. 2019, 105, 100577. [Google Scholar] [CrossRef]
- Favier, A.; De Wolf, C.; Scrivener, K.; Habert, G. A Sustainable Future for the European Cement and Concrete Industry: Technology Assessment for Full Decarbonisation of the Industry by 2050; ETH Zurich: Zurich, Switzerland, 2018. [Google Scholar] [CrossRef]
- You, S.; Ho, S.W.; Li, T.; Maneerung, T.; Wang, C.H. Techno-economic analysis of geopolymer production from the coal fly ash with high iron oxide and calcium oxide contents. J. Hazard. Mater. 2019, 361, 237–244. [Google Scholar] [CrossRef]
- Machiels, L.; Arnout, L.; Yan, P.; Jones, P.T.; Blanpain, B.; Pontikes, Y. Transforming enhanced landfill mining derived gasification/vitrification glass into low-carbon inorganic polymer binders and building products. J. Sustain. Metal. 2017, 3, 405–415. [Google Scholar] [CrossRef]
- Provis, J.L.; Van Deventer, J.S. Alkali Activated Materials: State-of-the-Art Report; RILEM TC 224-AAM; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Cartwright, C.; Rajabipour, F.; Radlińska, A. Shrinkage characteristics of alkali-activated slag cements. J. Mater. Civ. Eng. 2014, 27, B4014007. [Google Scholar] [CrossRef]
- Lee, N.K.; Jang, J.G.; Lee, H.K. Shrinkage characteristics of alkali-activated fly ash/slag paste and mortar at early ages. Cement Concr. Comp. 2014, 53, 239–248. [Google Scholar] [CrossRef]
- Palacios, M.; Puertas, F. Effect of shrinkage-reducing admixtures on the properties of alkali-activated slag mortars and pastes. Cement Concr. Res. 2007, 37, 691–702. [Google Scholar] [CrossRef]
- Collins, F.; Sanjayan, J.G. Effect of pore size distribution on drying shrinking of alkali-activated slag concrete. Cement Concr. Res. 2000, 30, 1401–1406. [Google Scholar] [CrossRef]
- Mounanga, P.; Bouasker, M.; Pertue, A.; Perronnet, A.; Khelidj, A. Early-age autogenous cracking of cementitious matrices: Physico-chemical analysis and micro/macro investigations. Mater. Struct. 2011, 44, 749–772. [Google Scholar] [CrossRef]
- Beersaerts, G.; Pontikes, Y. An attempt to mitigate the drying shrinkage of inorganic polymers by testing reactive and non-reactive admixtures. Mater. Lett. 2019, in press. [Google Scholar]
- Peys, A.; White, C.E.; Rahier, H.; Blanpain, B.; Pontikes, Y. Alkali-activation of CaO-FeOx-SiO2 slag: Formation mechanism from in-situ X-ray total scattering. Cement Concr. Res. 2019, 122, 179–188. [Google Scholar] [CrossRef]
- Van Jaarsveld, J.; Van Deventer, J.; Lukey, G. The effect of composition and temperature on the properties of fly ash- and kaolinite-based geopolymers. Chem. Eng. J. 2002, 89, 63–73. [Google Scholar] [CrossRef]
- Bakharev, T.; Sanjayan, J.G.; Cheng, Y.B. Effect of elevated temperature curing on properties of alkali-activated slag concrete. Cement Concr. Res. 1999, 29, 1619–1625. [Google Scholar] [CrossRef]
- Onisei, S.; Lesage, K.; Blanpain, B.; Pontikes, Y. Early Age Microstructural Transformations of an Inorganic Polymer Made of Fayalite Slag. J. Am. Ceram. Soc. 2015, 98, 2269–2277. [Google Scholar] [CrossRef]
- Close, P.; Shepherd, H.M.; Drummond, C.H. Determination of several valences of iron, arsenic and antimony, and selenium in glass. J. Am. Ceram. Soc. 1958, 41, 455–460. [Google Scholar] [CrossRef]
- ISO 9277:2010. Determination of the Specific Surface Area of Solids by Gas Adsorption—BET Method. Available online: https://www.iso.org/standard/44941.html (accessed on 1 November 2018).
- EN 196-6:2018. Methods of Testing Cement—Part 6: Determination of Fineness. Available online: https://shop.bsigroup.com/ProductDetail?pid=000000000030356711 (accessed on 1 November 2018).
- Zeng, Q.; Li, K.; Fen-Chong, T.; Dangla, P. Pore structure of cement pastes through NAD and MIP analysis. Adv. Cem. Res. 2015, 28, 23–32. [Google Scholar] [CrossRef]
- EN 196-1:2016. Methods of Testing Cement—Determination of Strength. Available online: http://store.uni.com/catalogo/en-196-1-2016 (accessed on 1 November 2018).
- ASTM C1698-09: 2014-Standard Test Method for Autogenous Strain of Cement Paste and Mortar; ASTM International: West Conshohocken, PA, USA, 2014.
- British Standards Institution. BS EN 12617–4:2002 Products and Systems for the Protection and Repair of Concrete Structures; Test Methods, Determination of Shrinkage and Expansion: Milton Keynes, UK, 2002; p. 14. [Google Scholar]
- Novais, R.M.; Ascensão, G.; Seabra, M.P.; Labrincha, J.A. Waste glass from end-of-life fluorescent lamps as raw material in geopolymers. Waste Manag. 2016, 52, 245–255. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Provis, J.L.; Bullen, F.; Reid, A.; Zhu, Y. Quantitative kinetic and structural analysis of geopolymers. Part 1. The activation of metakaolin with sodium hydroxide. Thermochim. Acta 2012, 539, 23–33. [Google Scholar] [CrossRef]
- Zhang, Z.; Provis, J.L.; Wang, H.; Bullen, F.; Reid, A. Quantitative kinetic and structural analysis of geopolymers. Part 2. Thermodynamics of sodium silicate activation of metakaolin. Thermochim. Acta 2013, 565, 163–171. [Google Scholar] [CrossRef]
- Van Deventer, J.S.J.; Provis, J.L.; Duxson, P.; Lukey, G.C. Reaction mechanisms in the geopolymeric conversion of inorganic waste to useful products. J. Hazard. Mater. 2007, 139, 506–513. [Google Scholar] [CrossRef]
- Ascensão, G.; Marchi, M.; Segata, M.; Faleschini, F.; Pontikes, Y. Increasing the dimensional stability of CaO-FeOx-Al2O3-SiO2 alkali-activated materials: A study on the swelling potential of calcium oxide-rich admixtures. Detritus J. 2019, in press. [Google Scholar]
- Salman, M.; Cizer, Ö.; Pontikes, Y.; Snellings, R.; Vandewalle, L.; Blanpain, B.; Van Balen, K. Cementitious binders from activated stainless steel refining slag and the effect of alkali solutions. J. Hazard. Mater. 2015, 286, 211–219. [Google Scholar] [CrossRef]
- Daux, V.; Guy, C.; Advocat, T.; Crovisier, J.L.; Stille, P. Kinetic aspects of basaltic glass dissolution at 90 C: Role of aqueous silicon and aluminium. Chem. Geol. 1997, 142, 109–126. [Google Scholar] [CrossRef]
- He, P.; Wang, M.; Fu, S.; Jia, D.; Yan, S.; Yuan, J.; Zhou, Y. Effects of Si/Al ratio on the structure and properties of metakaolin based geopolymer. Ceram. Int. 2016, 42, 14416–14422. [Google Scholar] [CrossRef]
- Kriskova, L.; Machiels, L.; Pontikes, Y. Inorganic polymers from a plasma convertor slag: Effect of activating solution on microstructure and properties. J. Sustain. Metal. 2015, 1, 240–251. [Google Scholar] [CrossRef]
- Hertel, T.; Blanpain, B.; Pontikes, Y. A proposal for a 100% use of bauxite residue towards inorganic polymer mortar. J. Sustain. Metal. 2016, 2, 394–404. [Google Scholar] [CrossRef]
- Dalby, K.N.; King, P.L. A new approach to determine and quantify structural units in silicate glasses using micro-reflectance Fourier-Transform infrared spectroscopy. Am. Miner. 2006, 91, 1783–1793. [Google Scholar] [CrossRef]
- Soliman, A.M.; Nehdi, M.L. Effects of shrinkage reducing admixture and wollastonite microfiber on early-age behavior of ultra-high performance concrete. Cement Concr. Comp. 2014, 46, 81–89. [Google Scholar] [CrossRef]
- Sant, G.; Lothenbach, B.; Juilland, P.; Le Saout, G.; Weiss, J.; Scrivener, K. The origin of early age expansions induced in cementitious materials containing shrinkage reducing admixtures. Cement Concr. Res. 2011, 41, 218–229. [Google Scholar] [CrossRef]
- Bumanis, G.; Vitola, L.; Bajare, D.; Dembovska, L.; Pundiene, I. Impact of reactive SiO2/Al2O3 ratio in precursor on durability of porous alkali activated materials. Ceram. Int. 2017, 43, 5471–5477. [Google Scholar] [CrossRef]
- Kalina, L.; Bílek, V.; Bartoníčková, E.; Krouská, J. Polypropylene Glycols as Effective Shrinkage-Reducing Admixtures in Alkali-Activated Materials. ACI Mater. J. 2018, 115, 51–256. [Google Scholar] [CrossRef]
- Thomas, R.J.; Lezama, D.; Peethamparan, S. On drying shrinkage in alkali-activated concrete: Improving dimensional stability by aging or heat-curing. Cement Concr. Res. 2017, 91, 13–23. [Google Scholar] [CrossRef]
- Ascensão, G.; Faleschini, F.; Vleugels, J.; Rahier, H.; Marchi, M.; Segata, M.; Faleschini, F.; Pontikes, Y. The effect of polypropylene glycols on the properties of Fe-rich alkali activated materials. In Proceedings of the 17th International Waste Management and Landfill Symposium, Cagliari, Italy, 30 September–4 October 2019. [Google Scholar]
- Kuenzel, C.; Vandeperre, L.J.; Donatello, S.; Boccaccini, A.R.; Cheeseman, C. Ambient temperature drying shrinkage and cracking in metakaolin-based geopolymers. J. Am. Ceram. Soc. 2012, 95, 3270–3277. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Mixture Portion (wt.%) | Slag/liquid | Solid/liquid | |||
|---|---|---|---|---|---|---|
| PS | KO | Solution (aq.) | Agg. | (g/g) | (g/g) | |
| PS mortars | 34.72 | - | 13.20 | 52.08 | 2.63 | 6.57 |
| KO mortars | - | 34.72 | 13.20 | 52.08 | ||
| Formulation | Binders Molar Ratios | ||
|---|---|---|---|
| SiO2/Al2O3 | M2O/SiO2 | H2O/M2O | |
| PS pastes | 4.45 | 0.08 | 25.71 |
| KO pastes | 6.07 | 0.12 | 18.25 |
| Oxide (wt.%) | PSslag | KOslag |
|---|---|---|
| Na2O | 0.3 | 2.0 |
| MgO | 1.3 | 1.5 |
| Al2O3 | 16.2 | 10.4 |
| SiO2 | 34.9 | 34.8 |
| P2O5 | 0.1 | 1.0 |
| SO3 | 0.0 | 0.6 |
| K2O | 0.5 | 0.2 |
| CaO | 23.1 | 3.3 |
| TiO2 | 0.6 | 0.3 |
| MnO | 0.1 | 0.9 |
| FeOx | 22.8 | 43.6 |
| Loss on ignition | 1.9 | 4.1 |
| Fe3+/Fetotal | 0.08 | 0.06 |
| Code | Density | Blaine | T-Plot |
|---|---|---|---|
| (g/cm3) | (cm2/g) | (m2/g) | |
| PS slag | 2.97 | 4500 ± 200 | 0.55 |
| KO slag | 3.41 | 5500 ± 400 | 1.01 |
| Code | Curing Cond | Peak Band I | Peak Band II | Peak Band III |
|---|---|---|---|---|
| (cm−1) | (cm−1) | (cm−1) | ||
| PS | Room | 448 | 712 | 970 |
| Heat | 431 | 693 | 948 | |
| Saturated | 439 | 699 | 976 | |
| KO | Room | 443 | 695 | 969 |
| Heat | 450 | 695 | 970 | |
| Saturated | 448 | 693 | 983 |
| Code | Curing Cond | Bulk Density | |||
|---|---|---|---|---|---|
| 1st day | 4th day | 28th day | 56th day | ||
| (g/cm3) | (g/cm3) | (g/cm3) | (g/cm3) | ||
| PS | Room | 2.37 ± 0.02 | 2.29 ± 0.01 | 2.26 ± 0.01 | 2.25 ± 0.01 |
| Heat | 2.39 ± 0.01 | 2.34 ± 0.01 | 2.34 ± 0.02 | 2.33 ± 0.02 | |
| Saturated | 2.38 ± 0.01 | 2.38 ± 0.01 | 2.38 ± 0.02 | 2.38 ± 0.02 | |
| KO | Room | 2.39 ± 0.02 | 2.33 ± 0.02 | 2.29 ± 0.02 | 2.29 ± 0.02 |
| Heat | 2.39 ± 0.01 | 2.35 ± 0.00 | 2.34 ± 0.02 | 2.34 ± 0.01 | |
| Saturated | 2.34 ± 0.01 | 2.34 ± 0.01 | 2.36 ± 0.01 | 2.33 ± 0.03 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ascensão, G.; Beersaerts, G.; Marchi, M.; Segata, M.; Faleschini, F.; Pontikes, Y. Shrinkage and Mitigation Strategies to Improve the Dimensional Stability of CaO-FeOx-Al2O3-SiO2 Inorganic Polymers. Materials 2019, 12, 3679. https://doi.org/10.3390/ma12223679
Ascensão G, Beersaerts G, Marchi M, Segata M, Faleschini F, Pontikes Y. Shrinkage and Mitigation Strategies to Improve the Dimensional Stability of CaO-FeOx-Al2O3-SiO2 Inorganic Polymers. Materials. 2019; 12(22):3679. https://doi.org/10.3390/ma12223679
Chicago/Turabian StyleAscensão, Guilherme, Glenn Beersaerts, Maurizio Marchi, Monica Segata, Flora Faleschini, and Yiannis Pontikes. 2019. "Shrinkage and Mitigation Strategies to Improve the Dimensional Stability of CaO-FeOx-Al2O3-SiO2 Inorganic Polymers" Materials 12, no. 22: 3679. https://doi.org/10.3390/ma12223679
APA StyleAscensão, G., Beersaerts, G., Marchi, M., Segata, M., Faleschini, F., & Pontikes, Y. (2019). Shrinkage and Mitigation Strategies to Improve the Dimensional Stability of CaO-FeOx-Al2O3-SiO2 Inorganic Polymers. Materials, 12(22), 3679. https://doi.org/10.3390/ma12223679