Electronic Structure of B20 (FeSi-Type) Transition-Metal Monosilicides



Abstract
1. Introduction
2. Calculation Methods
3. Results of Band Structure Calculations
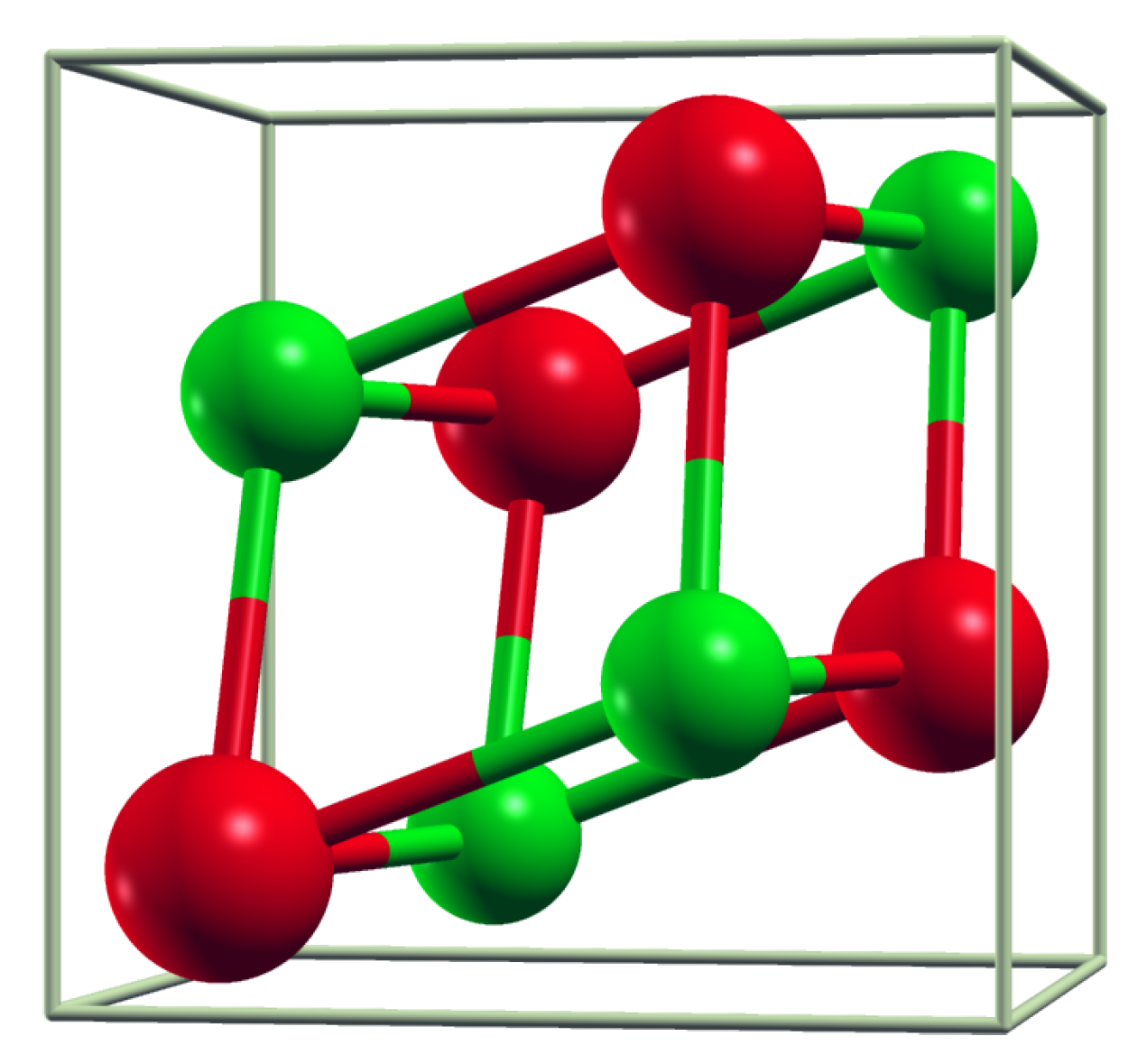
3.1. Crystal Structure Parameters
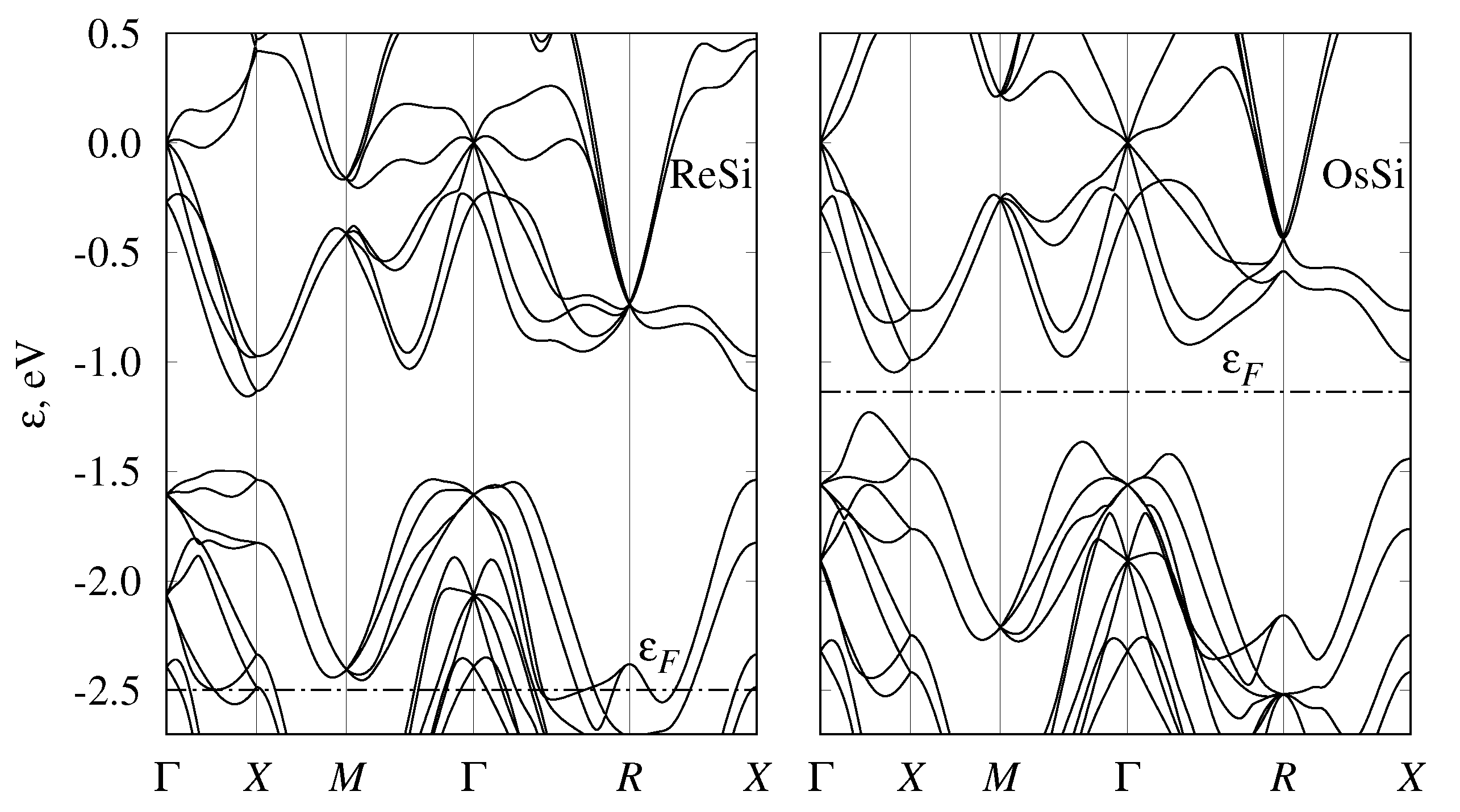
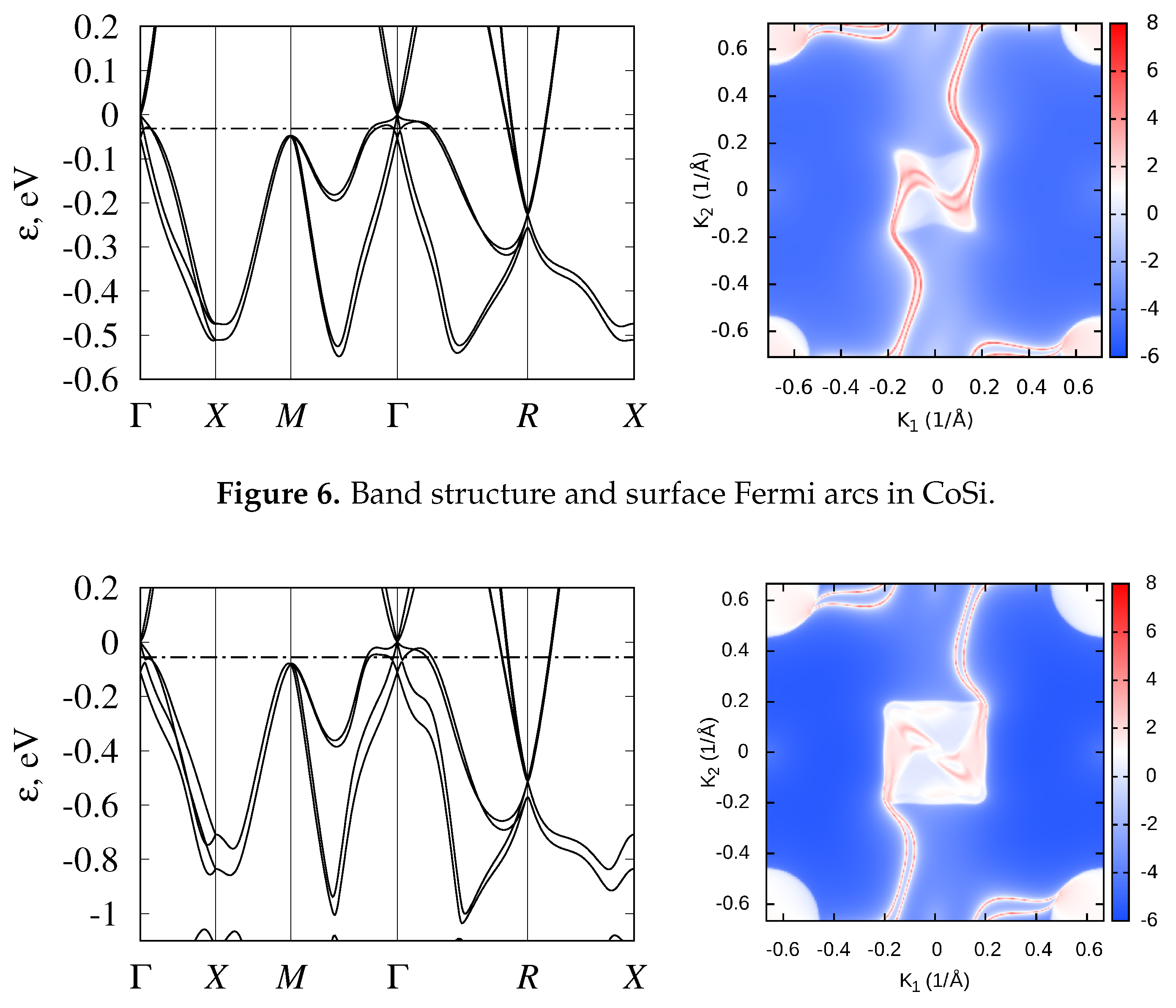
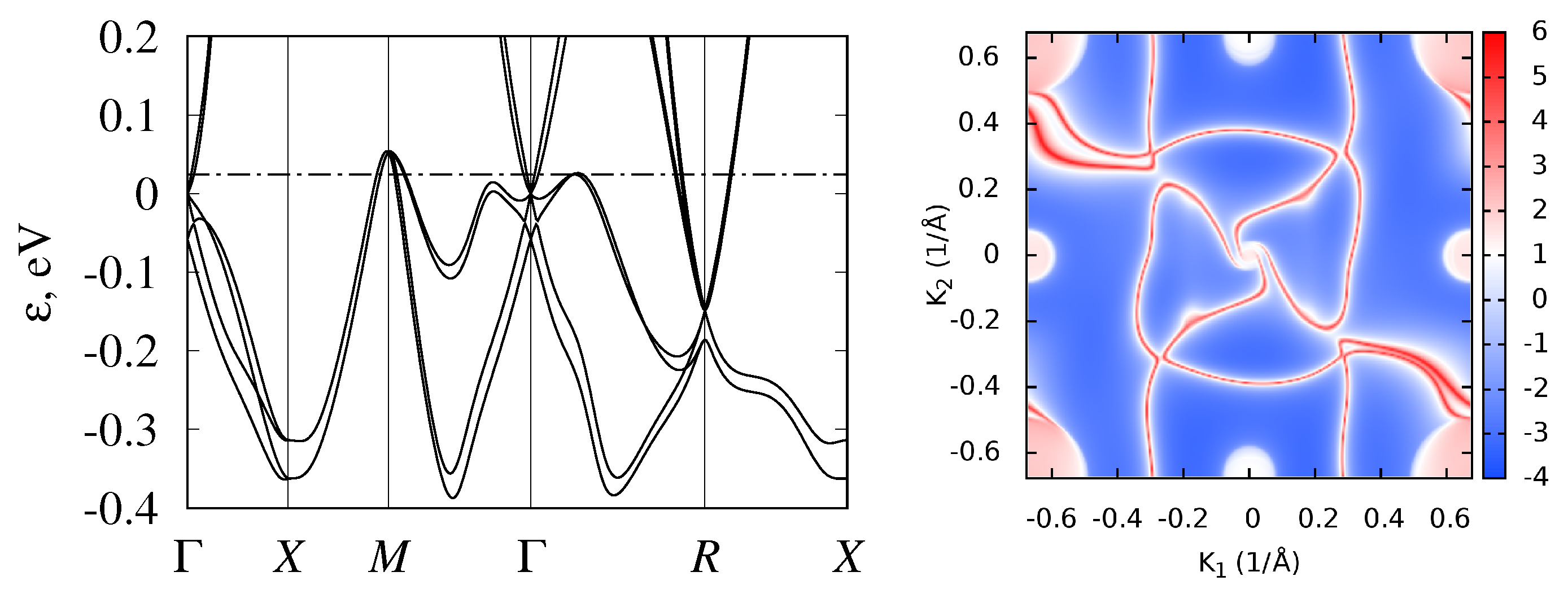
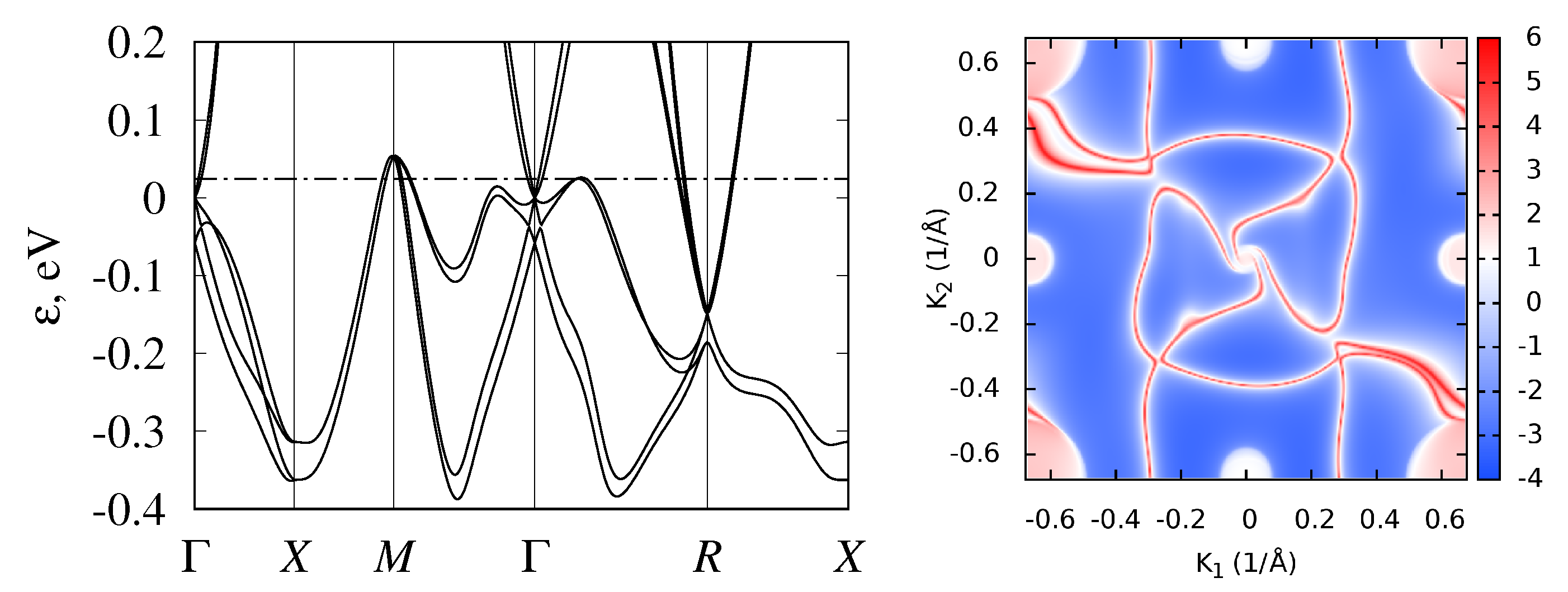
3.2. Band Structure and Topological Properties of Semimetallic Co and Rh Monosilicides and Monogermanides, and of RhSn
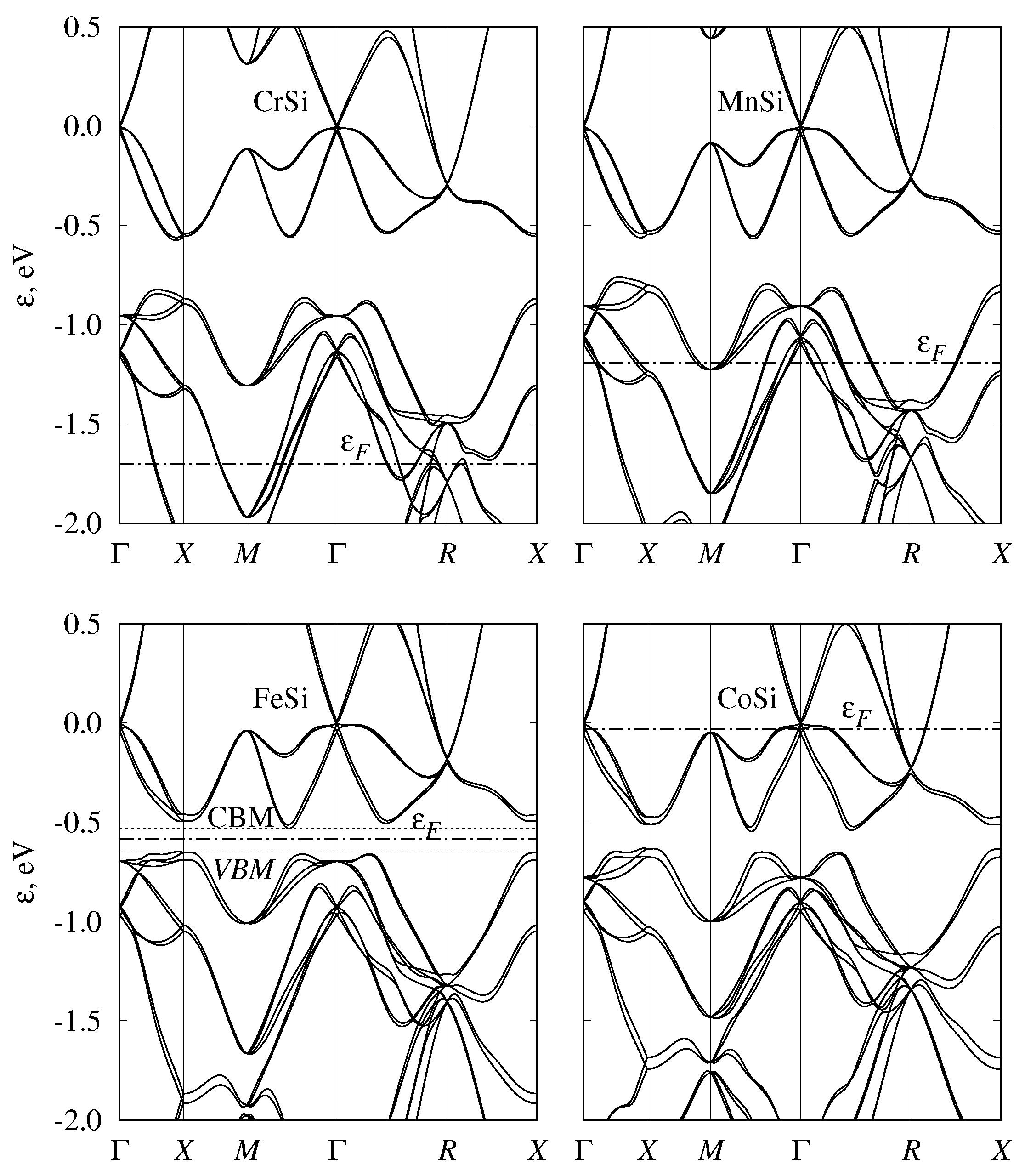
3.3. Semiconducting-Monosilicide Band Structure
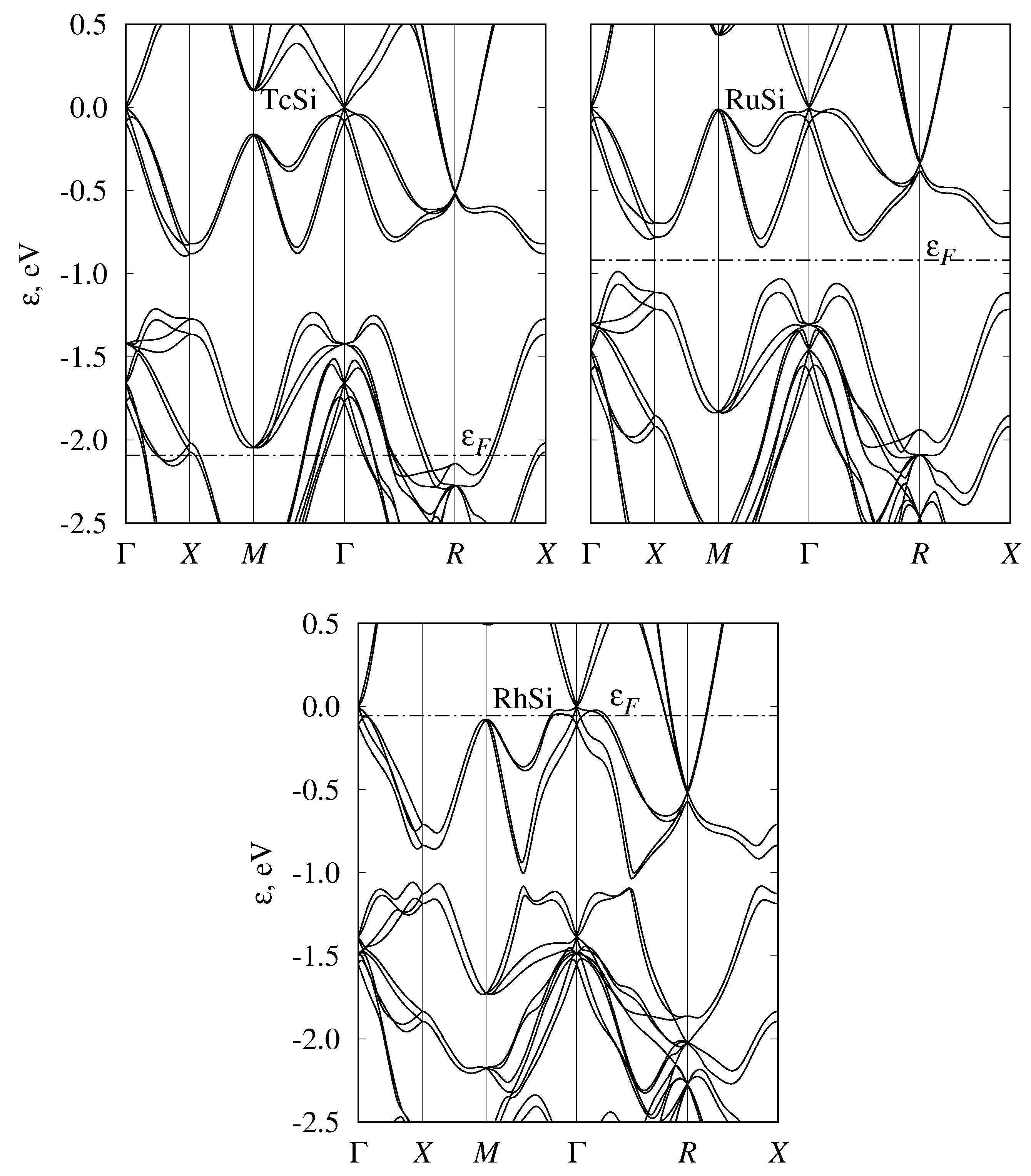
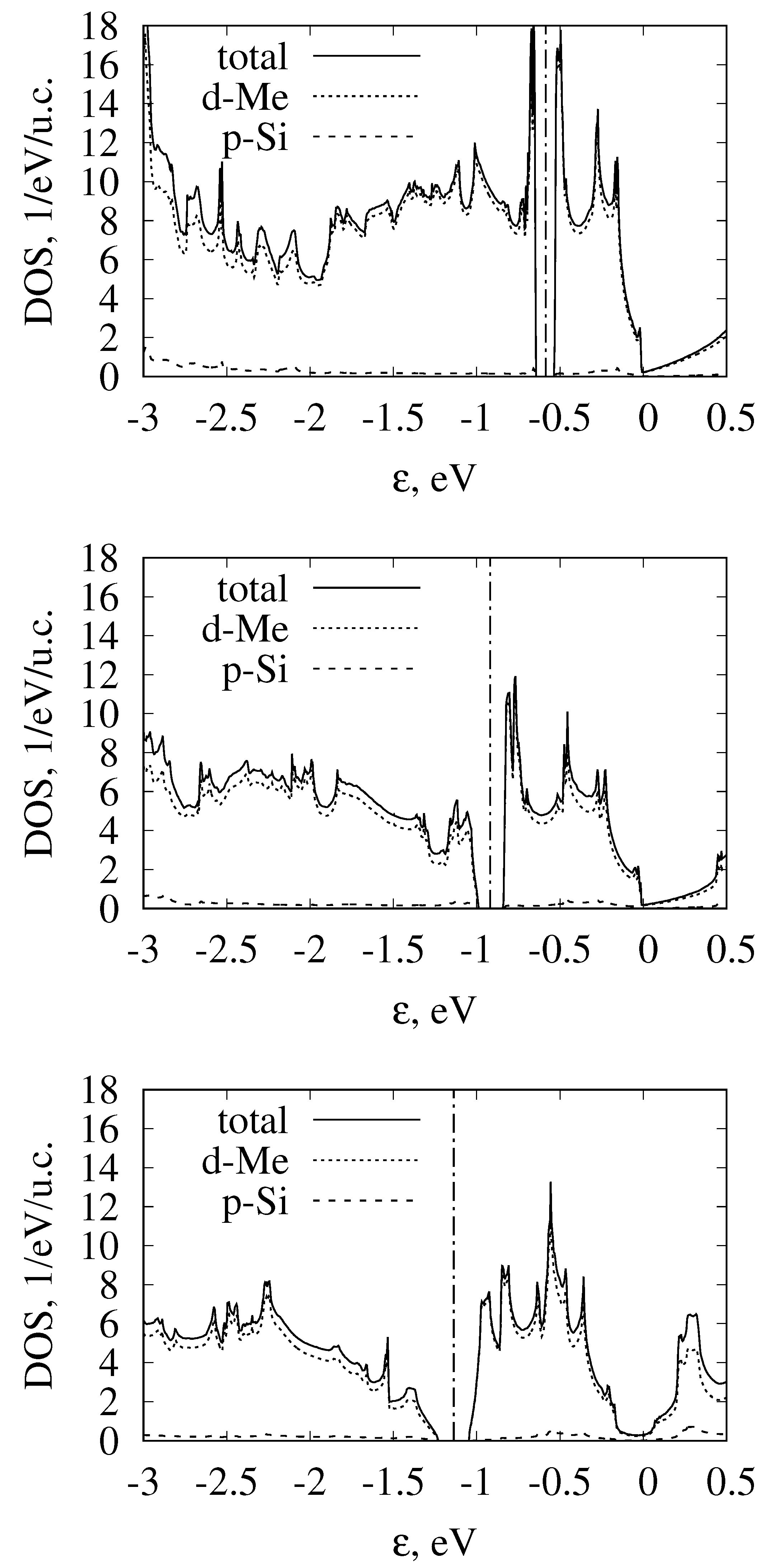
3.4. Band Structure of Metallic Monosilicides
4. Influence of Band Structure Peculiarities on Monosilicide Thermoelectric Properties
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mühlbauer, S.; Binz, B.; Jonietz, F.; Pfleiderer, C.; Rosch, A.; Neubauer, A.; Georgii, R.; Böni, P. Skyrmion Lattice in a Chiral Magnet. Science 2009, 323, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, N. Charge and Heat Transport Phenomena in Electronic and Spin Structures in B20-Type Compounds; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Bradlyn, B.; Cano, J.; Wang, Z.; Vergniory, M.G.; Felser, C.; Cava, R.J.; Bernevig, B.A. Beyond Dirac and Weyl fermions: Unconventional quasiparticles in conventional crystals. Science 2016, 353, 558. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Zhou, Q.; Zhang, S.C. Multiple Types of Topological Fermions in Transition Mmetal Silicides. Phys. Rev. Lett. 2017, 119, 206402. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Xu, S.Y.; Wieder, B.J.; Huang, D.S.S.S.M.; Belopolski, I.; Chang, T.R.; Zhang, S.; Bansil, A.; Lin, H.; Hasan, M.Z. Unconventional Chiral Fermions and Large Topological Fermi Arcs in RhSi. Phys. Rev. Lett. 2017, 119, 206401. [Google Scholar] [CrossRef] [PubMed]
- Pshenay-Severin, D.A.; Ivanov, Y.V.; Burkov, A.A.; Burkov, A.T. Band structure and unconventional electronic topology of CoSi. J. Phys. Condens. Matter 2018, 30, 135501. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, M.I.; Zaitsev, V.K. Semimetals as materials for thermoelectric generators. In CRC Handbook of Thermoelectrics; Rowe, D.M., Ed.; CRC Press: Boca Raton, FL, USA, 1995; Chapter 27. [Google Scholar]
- Asanabe, S.; Shinoda, D.; Sasaki, Y. Semimetallic Properties of Co1−xFexSi Solid Solutions. Phys. Rev. 1964, 134, A774. [Google Scholar] [CrossRef]
- Pan, Z.; Zhang, L.; Wu, J. Electronic structure and transport properties of doped CoSi single crystal. J. Appl. Phys. 2007, 101, 033715. [Google Scholar] [CrossRef]
- Sakai, A.; Ishii, F.; Onose, Y.; Tomioka, Y.; Yotsuhashi, S.; Adachi, H.; Nagaosa, N.; Tokura, Y. Thermoelectric power in transition-metal monosilicides. J. Phys. Soc. Jpn. 2007, 76, 093601. [Google Scholar] [CrossRef]
- Ishii, F.; Kotaka, H.; Onishi, T. Spin–Orbit Interaction Effects in the Electronic Structure of B20-Type CoSi: First-Principles Density Functional Study. JPS Conf. Proc. 2014, 3, 016019. [Google Scholar]
- Altintas, B. A comparative study on electronic and structural properties of transition metal monosilicides, CrSi (B20-type), RhSi (B20-type), RhSi (B31-type) and RhSi (B2-type). J. Phys. Chem. Solids 2011, 72, 1325–1329. [Google Scholar] [CrossRef]
- Niranjan, M.K. First principles study of structural, electronic and elastic properties of cubic and orthorhombic RhSi. Intermetallics 2012, 26, 150–156. [Google Scholar] [CrossRef]
- Takane, D.; Wang, Z.; Souma, S.; Nakayama, K.; Nakamura, T.; Oinuma, H.; Nakata, Y.; Iwasawa, H.; Cacho, C.; Kim, T.; et al. Observation of Chiral Fermions with a Large Topological Charge and Associated Fermi-Arc Surface States in CoSi. Phys. Rev. Lett. 2019, 122, 076402. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.; Li, H.; Zhang, T.; Tian, S.; Li, C.; Fu, B.; Tang, C.; Wang, L.; Li, Z.; Fan, W.; et al. Observation of unconventional chiral fermions with long Fermi arcs in CoSi. Nature 2019, 567, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.S.; Belopolski, I.; Cochran, T.A.; Xu, X.; Yin, J.X.; Chang, G.; Xie, W.; Manna, K.; Süß, V.; Huang, C.Y.; et al. Topological chiral crystals with helicoid-arc quantum states. Nature 2019, 567, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, N.; Onose, Y.; Shiomi, Y.; Ishiwata, S.; Tokura, Y. Band-filling dependence of thermoelectric properties in B20-type CoGe. Appl. Phys. Lett. 2012, 100, 093902. [Google Scholar] [CrossRef]
- Tsvyashchenko, A.; Sidorov, V.; Petrova, A.; Fomicheva, L.; Zibrov, I.; Dmitrienko, V. Superconductivity and magnetism in noncentrosymmetric RhGe. J. Alloys Compd. 2016, 686, 431–437. [Google Scholar] [CrossRef]
- Magnitskaya, M.; Chtchelkatchev, N.; Tsvyashchenko, A.; Salamatin, D.; Lepeshkin, S.; Fomicheva, L.; Budzyński, M. Electron and phonon properties of noncentrosymmetric RhGe from ab initio calculations. J. Magn. Magn. Mater. 2019, 470, 127–130. [Google Scholar] [CrossRef]
- Bradlyn, B.; Elcoro, L.; Cano, J.; Vergniory, M.G.; Wang, Z.; Felser, C.; Aroyo, M.I.; Bernevig, B.A. Topological quantum chemistry. Nature 2017, 547, 298–305. [Google Scholar] [CrossRef]
- Vergniory, M.G.; Elcoro, L.; Felser, C.; Regnault, N.; Bernevig, B.A.; Wang, Z. A complete catalogue of high-quality topological materials. Nature 2019, 566, 480–485. [Google Scholar] [CrossRef]
- Topological Material Database. Available online: topologicalquantumchemistry.org (accessed on 15 August 2019).
- Zhao, Y.N.; Han, H.L.; Yu, Y.; Xue, W.H.; Gao, T. First-principles studies of the electronic and dynamical properties of monosilicides MSi (M = Fe, Ru, Os). EPL 2009, 85, 47005. [Google Scholar] [CrossRef]
- He, G.-M.; Li, S.-P.; Huang, M.-C. Structural and Electronic Properties of RuSi, RuGe and OsSi. Chin. Phys. Lett. 2001, 18, 1389–1391. [Google Scholar] [CrossRef]
- Minisini, B.; Roetting, J.; Tsobnang, F. Elastic and thermodynamic properties of OsSi, OsSi2 and Os2Si3. Comput. Mater. Sci. 2008, 43, 812–817. [Google Scholar] [CrossRef]
- Li, J.; Linghu, R.-F.; Yang, Z.-J.; Cao, Y.; Yang, X.-D. First-Principles Study of Structural, Elastic and Electronic Properties of OsSi. Commun. Theor. Phys. 2009, 52, 701. [Google Scholar]
- Wolfe, R.; Wernick, J.; Haszko, S. Thermoelectric properties of FeSi. Phys. Lett. 1965, 19, 449–450. [Google Scholar] [CrossRef]
- Schlesinger, Z.; Fisk, Z.; Zhang, H.T.; Maple, M.B.; DiTusa, J.; Aeppli, G. Unconventional charge gap formation in FeSi. Phys. Rev. Lett. 1993, 71, 1748–1751. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Kimura, S.i.; Kulatov, E.; Halilov, S.V.; Nanba, T.; Motokawa, M.; Sato, M.; Nagasaka, K. Optical Measurements and Band Calculations of FeSi. J. Phys. Soc. Jpn. 1994, 63, 4206–4212. [Google Scholar] [CrossRef]
- Buschinger, B.; Guth, W.; Weiden, M.; Geibel, C.; Steglich, F.; Vescoli, V.; Degiorgi, L.; Wassilew-Reul, C. RuSi: Metal-semiconductor transition by change of structure. J. Alloys Compd. 1997, 262–263, 238–242. [Google Scholar] [CrossRef]
- Hohl, H.; Ramirez, A.; Goldmann, C.; Ernst, G.; Bucher, E. Transport properties of RuSi, RuGe, OsSi, and quasi-binary alloys of these compounds. J. Alloys Compd. 1998, 278, 39–43. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Tajima, K.; Bloch, D.; Roth, M. Helical spin structure in manganese silicide MnSi. Solid State Commun. 1976, 19, 525–528. [Google Scholar] [CrossRef]
- Thessieu, C.; Flouquet, J.; Lapertot, G.; Stepanov, A.; Jaccard, D. Magnetism and spin fluctuations in a weak itinerant ferromagnet: MnSi. Solid State Commun. 1995, 95, 707–712. [Google Scholar] [CrossRef]
- Pfleiderer, C.; McMullan, G.J.; Julian, S.R.; Lonzarich, G.G. Magnetic quantum phase transition in MnSi under hydrostatic pressure. Phys. Rev. B 1997, 55, 8330–8338. [Google Scholar] [CrossRef]
- Nakanishi, O.; Yanase, A.; Hasegawa, A. Electronic energy band structure of MnSi. J. Magn. Magn. Mater. 1980, 15–18, 879–880. [Google Scholar] [CrossRef]
- Imai, Y.; Mukaida, M.; Kobayashi, K.; Tsunoda, T. Calculation of the density of states of transition metal monosilicides by a first-principle pseudopotential method using plane-wave basis. Intermetallics 2001, 9, 261–268. [Google Scholar] [CrossRef]
- Yamada, H.; Terao, K. Itinerant-electron metamagnetism of MnSi at high pressure. Phys. Rev. B 1999, 59, 9342–9347. [Google Scholar] [CrossRef]
- Jeong, T.; Pickett, W.E. Implications of the B20 crystal structure for the magnetoelectronic structure of MnSi. Phys. Rev. B 2004, 70, 075114. [Google Scholar] [CrossRef]
- Collyer, R.; Browne, D. Correlations and the Magnetic Moment of MnSi. Phys. B Condens. Matter 2008, 403, 1420–1422. [Google Scholar] [CrossRef]
- Shanavas, K.V.; Satpathy, S. Electronic structure and the origin of the Dzyaloshinskii-Moriya interaction in MnSi. Phys. Rev. B 2016, 93, 195101. [Google Scholar] [CrossRef]
- Bak, P.; Jensen, M.H. Theory of helical magnetic structures and phase transitions in MnSi and FeGe. J. Phys. C Solid State Phys. 1980, 13, L881–L885. [Google Scholar] [CrossRef]
- Povzner, A.; Volkov, A.; Nuretdinov, T.; Nogovitsyna, T. Magnetic Phase Transition in MnSi on the Base of the LSDA + U + SO Calculations of the Electronic Structure and the Spin-Fluctuation Theory. Phys. Solid State 2018, 60, 1924–1929. [Google Scholar] [CrossRef]
- Nicolaou, A.; Gatti, M.; Magnano, E.; Le Fèvre, P.; Bondino, F.; Bertran, F.; Tejeda, A.; Sauvage-Simkin, M.; Vlad, A.; Garreau, Y.; et al. Fermi surface symmetry and evolution of the electronic structure across the paramagnetic-helimagnetic transition in MnSi/Si(111). Phys. Rev. B 2015, 92, 081110. [Google Scholar] [CrossRef]
- Burkov, A.T. Silicide Thermoelectrics: Materials for Energy Harvesting. Phys. Status Solidi A 2018, 215, 1800105. [Google Scholar] [CrossRef]
- Franciosi, A.; Peterman, D.; Weaver, J.; Moruzzi, V. Structural morphology and electronic properties of the Si-Cr interface. Phys. Rev. B 1982, 25, 4981. [Google Scholar] [CrossRef]
- Kematick, R.; Myers, C. Band structures of and bonding in 10-electron solids: Aluminum-manganese (MnAl), chromium silicide (CrSi), vanadium phosphide (VP), titanium sulfide (TiS). Inorg. Chem. 1992, 31, 3568–3572. [Google Scholar] [CrossRef]
- Dutta, P.; Pandey, S.K. Investigating the electronic structure of MSi (M = Cr, Mn, Fe and Co) and calculating Ueff and J by using cDFT. Comput. Condens. Matter 2018, 16, e00325. [Google Scholar] [CrossRef]
- Alekseeva, G.T.; Zaitsev, V.K.; Petrov, A.V.; Tarasov, V.I.; Fedorov, M.I. Electrical properties and thermal conductivity of cobalt monosilicide and of solid solutions Co1−xFexSi and Co1−xNixSi. Sov. Phys. Solid State 1981, 23, 1685. [Google Scholar]
- Ou-Yang, T.; Zhuang, Y.; Ramachandran, B.; Chen, W.; Shu, G.; Hu, C.; Chou, F.; Kuo, Y. Effect of Co substitution on thermoelectric properties of FeSi. J. Alloys Compd. 2017, 702, 92–98. [Google Scholar] [CrossRef]
- Pshenay-Severin, D.A.; Ivanov, Y.V.; Burkov, A.T.; Novikov, S.V.; Zaitsev, V.K.; Reith, H. Electronic Structure and Thermoelectric Properties of Transition Metal Monosilicides. J. Electron. Mater. 2017. [Google Scholar] [CrossRef]
- Burkov, A.T.; Novikov, S.V.; Zaitsev, V.K.; Reith, H. Transport properties of cobalt monosilicide and its alloys at low temperatures. Semiconductors 2017, 51, 689–691. [Google Scholar] [CrossRef]
- Kuo, Y.K.; Sivakumar, K.M.; Huang, S.J.; Lue, C.S. Thermoelectric properties of the CoSi1−xGex alloys. J. Appl. Phys. 2005, 98, 123510. [Google Scholar] [CrossRef]
- Skoug, E.; Zhou, C.; Pei, Y.; Morelli, D.T. High thermoelectric power factor in alloys based on CoSi. Appl. Phys. Lett. 2009, 94, 022115. [Google Scholar] [CrossRef]
- Lue, C.S.; Kuo, Y.K.; Huang, C.L.; Lai, W.J. Hole-doping effect on the thermoelectric properties and electronic structure of CoSi. Phys. Rev. B 2004, 69, 125111. [Google Scholar] [CrossRef]
- Sun, H.; Morelli, D.T.; Kirkham, M.J.; Meyer, H.M.; Lara-Curzio, E. The role of boron segregation in enhanced thermoelectric power factor of CoSi1−xBx alloys. J. Appl. Phys. 2011, 110, 123711. [Google Scholar] [CrossRef]
- Sun, H.; Lu, X.; Morelli, D.T. Effects of Ni, Pd, and Pt Substitutions on Thermoelectric Properties of CoSi Alloys. J. Electron. Mater. 2013, 42, 1352–1357. [Google Scholar] [CrossRef]
- Sun, H.; Lu, X.; Morelli, D.T. Detrimental effect of powder processing on the thermoelectric properties of CoSi. J. Mater. Sci. 2017, 52, 8293–8299. [Google Scholar] [CrossRef]
- Sales, B.C.; Delaire, O.; McGuire, M.A.; May, A.F. Thermoelectric properties of Co-, Ir-, and Os-doped FeSi alloys: Evidence for strong electron-phonon coupling. Phys. Rev. B 2011, 83, 125209. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Mostofi, A.A.; Yates, J.R.; Pizzi, G.; Lee, Y.S.; Souza, I.; Vanderbilt, D.; Marzari, N. An updated version of wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 2014, 185, 2309–2310. [Google Scholar] [CrossRef]
- Kokalj, A. Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Comput. Mater. Sci. 2003, 28, 155–168. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, S.; Song, H.F.; Troyer, M.; Soluyanov, A.A. WannierTools: An open-source software package for novel topological materials. Comput. Phys. Commun. 2018, 224, 405. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Larchev, V.; Popova, S. The polymorphism of transition metal monogermanides at high pressures and temperatures. J. Less Common Met. 1982, 87, 53–57. [Google Scholar] [CrossRef]
- Mattheiss, L.F.; Hamann, D.R. Band structure and semiconducting properties of FeSi. Phys. Rev. B 1993, 47, 13114–13119. [Google Scholar] [CrossRef]
- Petrova, A.E.; Krasnorussky, V.N.; Yuhasz, W.M.; Lograsso, T.A.; Stishov, S.M. Elastic properties of MnSi, FeSi and CoSi. J. Phys. Conf. Ser. 2011, 273, 012056. [Google Scholar] [CrossRef]
- Franciosi, A.; Weaver, J.H.; O’Neill, D.G.; Schmidt, F.A.; Bisi, O.; Calandra, C. Electronic structure of Cr silicides and Si-Cr interface reactions. Phys. Rev. B 1983, 28, 7000–7008. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Brand, J. The constitution of the chromium-niobium-silicon system. J. Less Common Met. 1961, 3, 34–43. [Google Scholar] [CrossRef]
- Boren, B. Roentgenuntersuchung der Legierungen von Silicium mit Chrom, Mangan, Kobalt und Nickel. Arkiv Kemi Mineral Geol. 1933, 11A, 11. [Google Scholar]
- Pauling, L.; Soldate, A.M. The nature of the bonds in the iron silicide, FeSi, and related crystals. Acta Crystallogr. 1948, 1, 212–216. [Google Scholar] [CrossRef]
- Zelenin, L.P.; Sidorenko, F.A.; Geld, P.V. Structural peculiarities of ϵ-phase of Co-Si system. Izv. Vyss. Uchebnyh Zaved. Tsvetnaya Metall. 1964, 2, 146. (In Russian) [Google Scholar]
- Darby, J.; Downey, J.; Norton, L. Intermediate phases in the technetium-aluminum and technetium-silicon systems. J. Less Common Met. 1965, 8, 15–19. [Google Scholar] [CrossRef]
- Weitzer, F.; Perring, L.; Gachon, J.; Feschotte, P.; Schuster, J. Structure refinements of some compounds of the Ru-Si, Ru-Ge and Ru-Sn systems. Proc. Electrochem. Soc. 1997, 97, 241–249. [Google Scholar]
- Geller, S.; Wood, E. The Crystal Structure of Rhodium Silicide, RhSi. Acta Crystallogr. 1954, 7, 441–443. [Google Scholar] [CrossRef]
- Engstrom, I.; Johnsson, T. Least-squares Refinement of the Structure of RhSi (FeSi-type). Acta Chem. Scand. 1965, 19, 1508–1509. [Google Scholar] [CrossRef][Green Version]
- Fedyna, V.; Gladyshevskii, R. Refinement of the crystal structure of the compound ReSi. Visn. L’vivs’kogo (Derzhavnogo) Univ. Seriya Khimichna 2014, 55, 87–92. [Google Scholar] [CrossRef][Green Version]
- Korst, W.L.; Finnie, L.N.; Searcy, A.W. The Crystal Structures of the Monosilicides of Osmium, Iridium and Ruthenium. J. Phys. Chem. 1957, 61, 1541–1543. [Google Scholar] [CrossRef]
- Schubert, V.K. Untersuchungen im System Rh-Sn. Zeitschrift für Naturforschung. Teil A Astrophys. Phys. Phys. Chem. 1947, 36, 246. [Google Scholar]
- Huang, S.M.; Xu, S.Y.; Belopolski, I.; Lee, C.C.; Chang, G.; Chang, T.R.; Wang, B.; Alidoust, N.; Bian, G.; Neupane, M.; et al. New type of Weyl semimetal with quadratic double Weyl fermions. Proc. Natl. Acad. Sci. USA 2016, 113, 1180–1185. [Google Scholar] [CrossRef]
- Elcoro, L.; Bradlyn, B.; Wang, Z.; Vergniory, M.G.; Cano, J.; Felser, C.; Bernevig, B.A.; Orobengoa, D.; Flor, G.; Aroyo, M.I. Double crystallographic groups and their representations on the Bilbao Crystallographic Server. J. Appl. Crystallogr. 2017, 50, 1457–1477. [Google Scholar] [CrossRef]
- Shaposhnikov, V.L.; Migas, D.B.; Borisenko, V.E.; Dorozhkin, N.N. Features of the band structure for semiconducting iron, ruthenium, and osmium monosilicides. Semiconductors 2009, 43, 142–144. [Google Scholar] [CrossRef]
- Neef, M.; Doll, K.; Zwicknagl, G. Structural, electronic, and magnetic properties of FeSi: Hybrid functionals and non-local exchange. J. Phys. Condens. Matter 2006, 18, 7437–7447. [Google Scholar] [CrossRef]
- Mazurenko, V.V.; Shorikov, A.O.; Lukoyanov, A.V.; Kharlov, K.; Gorelov, E.; Lichtenstein, A.I.; Anisimov, V.I. Metal-insulator transitions and magnetism in correlated band insulators: FeSi and Fe1−xCoxSi. Phys. Rev. B 2010, 81, 125131. [Google Scholar] [CrossRef]
- Tomczak, J.M.; Haule, K.; Kotliar, G. Signatures of electronic correlations in iron silicide. Proc. Natl. Acad. Sci. USA 2012, 109, 3243–3246. [Google Scholar] [CrossRef]
- Dutta, P.; Pandey, S.K. Effects of correlations and temperature on the electronic structures and related physical properties of FeSi and CoSi: A comprehensive study. J. Phys. Condens. Matter 2019, 31, 145602. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Ezhov, S.Y.; Elfimov, I.S.; Solovyev, I.V.; Rice, T.M. Singlet Semiconductor to Ferromagnetic Metal Transition in FeSi. Phys. Rev. Lett. 1996, 76, 1735–1738. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Pshenay-Severin, D.A.; Ivanov, Y.V.; Burkov, A.T. The effect of energy-dependent electron scattering on thermoelectric transport in novel topological semimetal. J. Phys. Condens. Matter 2018, 30, 475501. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | , eV/at | , eV/at | Decomposes into |
|---|---|---|---|
| CrSi | 0.069 | CrSi(Pmn) + CrSi(I4/mmm) | |
| NiSi | 0.101 | NiSi(Pnma) | |
| RhSi | 0.032 | RhSi(Pnma) | |
| OsSi | 0.012 | SiOs(Pbcn) + Os(P6/mmc) | |
| IrSi | - | - | IrSi(Pnma) |
| CoGe | 0.006 | CoGe(C2/m) | |
| CoSn | - | - | CoSn(P6/mmm) |
| RhGe | 0.040 | RhGe(Pnma) |
| Material | , Å | , GPa | ||
|---|---|---|---|---|
| CrSi (0.48 /at) | 4.5895 (4.5947) | 0.1377 (0.1379) | 0.8477 (0.8473) | 207 (196) |
| 4.607 [69], 4.629 [70] | ||||
| MnSi (1 /at) | 4.4974 (4.5180) | 0.1374 (0.1366) | 0.8456 (0.8452) | 221 (210) |
| 4.5598 [68], 4.557 [71] | 0.138 [71] | 0.846 [71] | 164 [68] | |
| FeSi | 4.4489 | 0.1363 | 0.8408, | 219 |
| 4.483 [68], 4.489 [72] | 0.137 [72] | 0.842 [72] | 186 [68] | |
| CoSi | 4.4302 | 0.1451 | 0.8432 | 219 |
| 4.444 [68], 4.4445 [73] | 0.144 [73] | 0.846 [73] | 207 [68] | |
| TcSi | 4.7722 | 0.1348 | 0.8448 | 229 |
| 4.755 [74] | ||||
| RuSi | 4.7361 | 0.1295 | 0.8376 | 209 |
| 4.7058 [31], 4.7059 [75] | 0.1365 [31], 0.1302 [75] | 0.8426 [31], 0.8387 [75] | ||
| RhSi | 4.7199 | 0.1523 | 0.8419 | 222 |
| 4.6750 [76], 4.6740 [77] | 0.1440 [76], 0.1459 [77] | 0.8400 [76], 0.8403 [77] | ||
| ReSi | 4.8021 | 0.1343 | 0.8443 | 248 |
| 4.7744 [78] | 0.1346 [78] | 0.8375 [78] | ||
| OsSi | 4.7927 | 0.1234 | 0.8357 | 209 |
| 4.7290 [79] | ||||
| CoGe | 4.6424 | 0.1360 | 0.8394 | 155 |
| 4.637 [66] | ||||
| RhGe | 4.9405 | 0.1211 | 0.8351 | 98 |
| 4.862 [66] | ||||
| RhSn | 5.1986 | 0.1430 | 0.8397 | 141 |
| 5.122 [80] |
| Material | , eV | , eV | , eV | , eV |
|---|---|---|---|---|
| CoSi | ||||
| RhSi | ||||
| CoGe | 0.053 | |||
| RhGe | 0.347 | |||
| RhSn | 0.194 |
| Material | (SOC), eV | (without SOC), eV | (expt.), eV |
|---|---|---|---|
| FeSi | 0.111 | 0.196, 0.17 [83], 0.13 [23] | 0.054 [29], 0.055 [27], 0.06 [28] |
| RuSi | 0.146 | 0.235, 0.22 [83], 0.48 [24], 0.22 [23] | 0.26 [31], 0.2–0.3 [30] |
| OsSi | 0.182 | 0.518, 0.74 [24], 0.68 [26], 0.53 [23], 0.50 [83] | >0.26 [31] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pshenay-Severin, D.A.; Burkov, A.T. Electronic Structure of B20 (FeSi-Type) Transition-Metal Monosilicides. Materials 2019, 12, 2710. https://doi.org/10.3390/ma12172710
Pshenay-Severin DA, Burkov AT. Electronic Structure of B20 (FeSi-Type) Transition-Metal Monosilicides. Materials. 2019; 12(17):2710. https://doi.org/10.3390/ma12172710
Chicago/Turabian StylePshenay-Severin, Dmitry A., and Alexander T. Burkov. 2019. "Electronic Structure of B20 (FeSi-Type) Transition-Metal Monosilicides" Materials 12, no. 17: 2710. https://doi.org/10.3390/ma12172710
APA StylePshenay-Severin, D. A., & Burkov, A. T. (2019). Electronic Structure of B20 (FeSi-Type) Transition-Metal Monosilicides. Materials, 12(17), 2710. https://doi.org/10.3390/ma12172710