Comparison of Thermoresponsive Hydrogels Synthesized by Conventional Free Radical and RAFT Polymerization


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterizations
2.3. Experimental
2.3.1. Free Radical Polymerization (FRP) of NIPAAm
2.3.2. RAFT Polymerization of NIPAAm
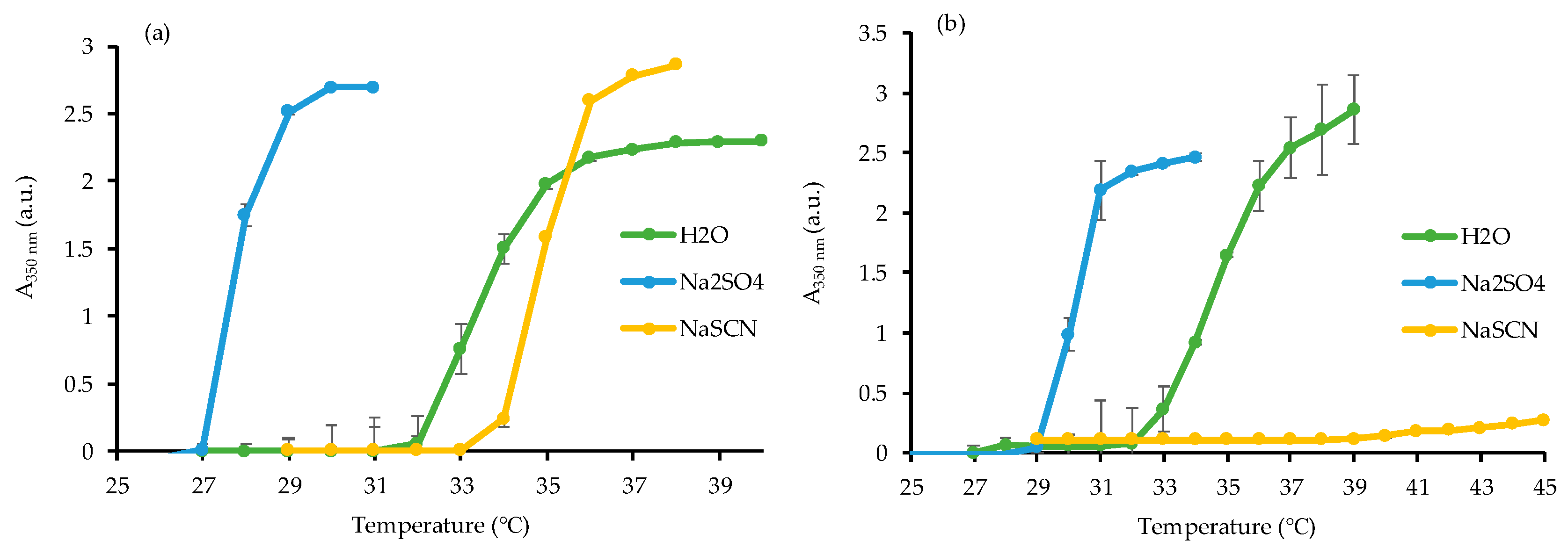
2.3.3. Determination of the Lower Critical Solution Temperature (LCST)
2.3.4. Preparation of PNIPAAm Gels via FRP
2.3.5. Preparation of PNIPAAm Gels via RAFT Polymerization
2.3.6. Scanning Electron Microscopy (SEM)
2.3.7. Swelling Testing
2.3.8. Determination of the Volume Phase Transition Temperature (VPTT)
2.3.9. Stability Study
2.3.10. Release Study of Fluorescein
3. Results and Discussion
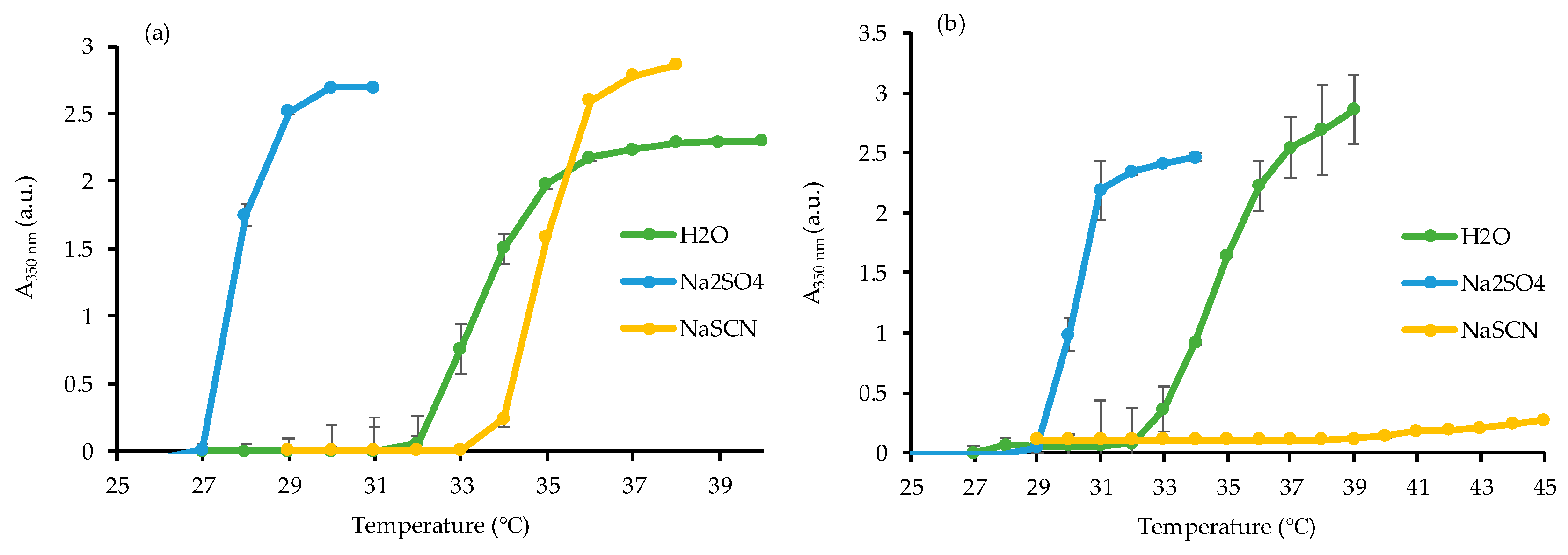
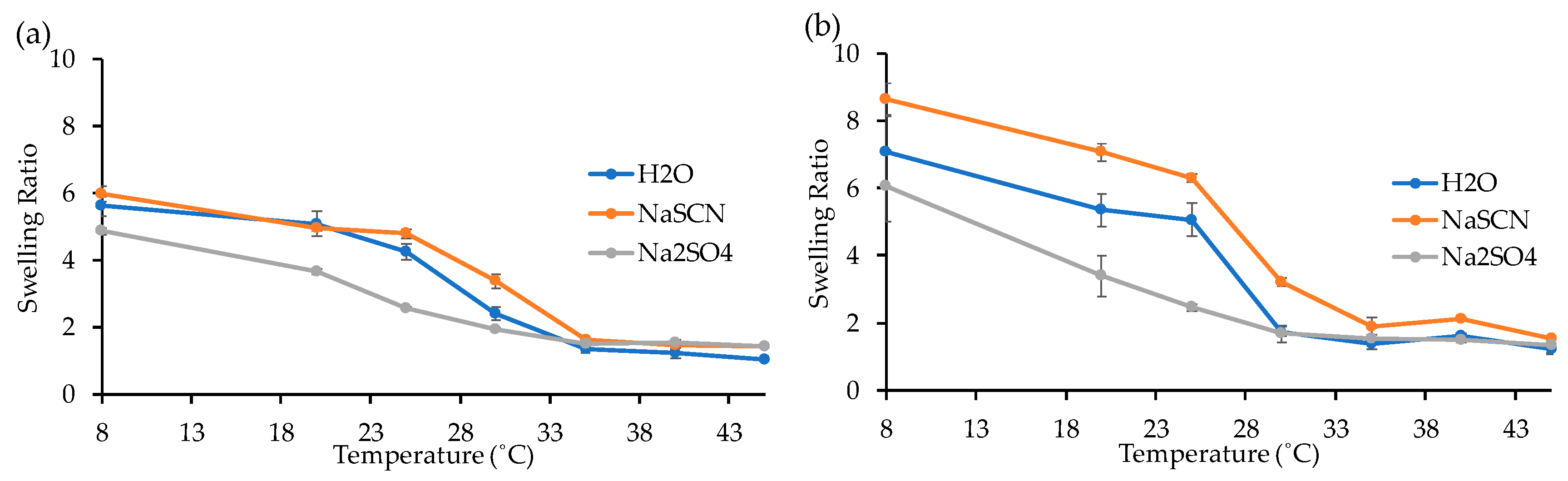
3.1. Synthesis of Homopolymer PNIPAAm and Study of Its LCST
3.2. PNIPAAm Gels
3.2.1. Preparation
3.2.2. SEM Imaging
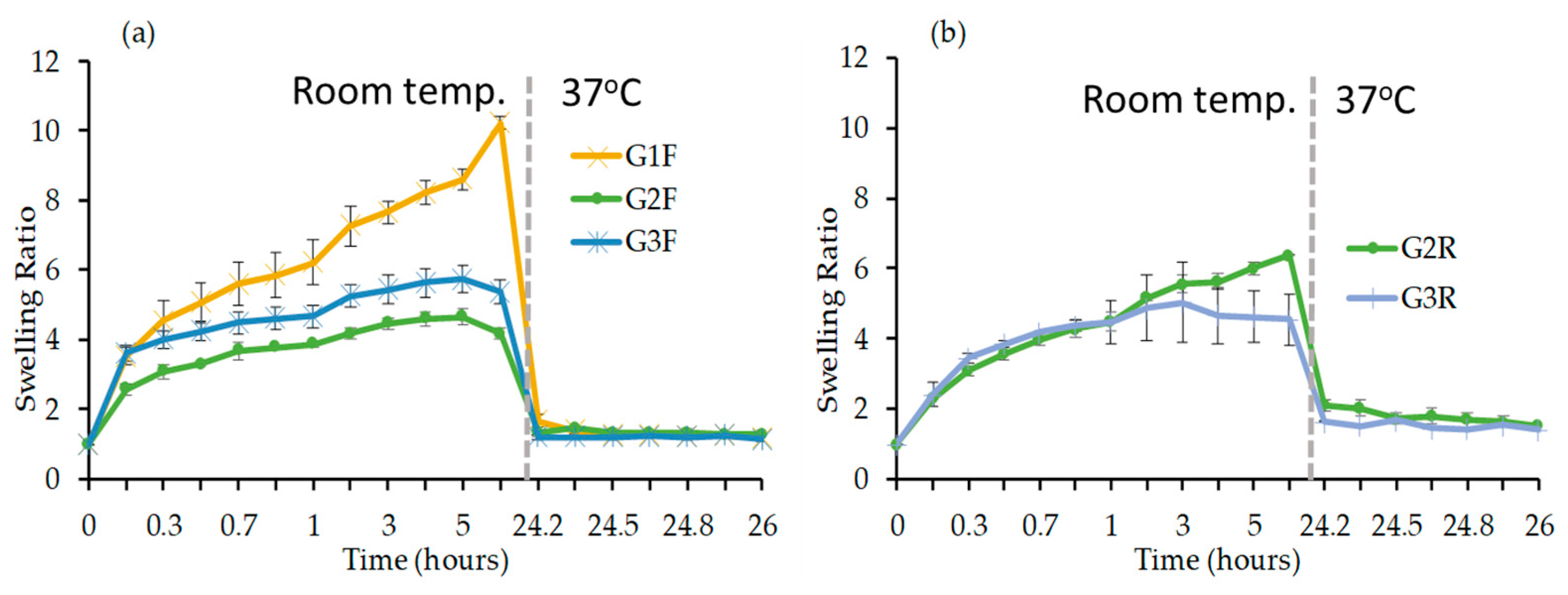
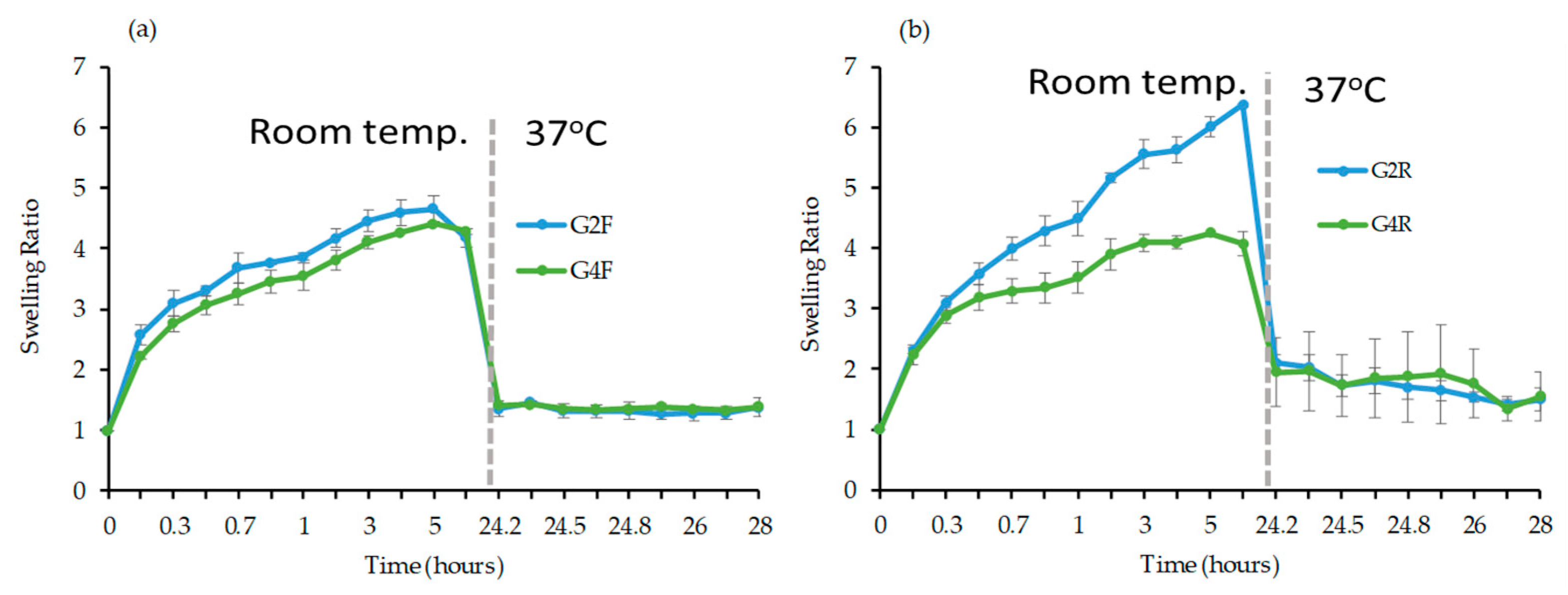
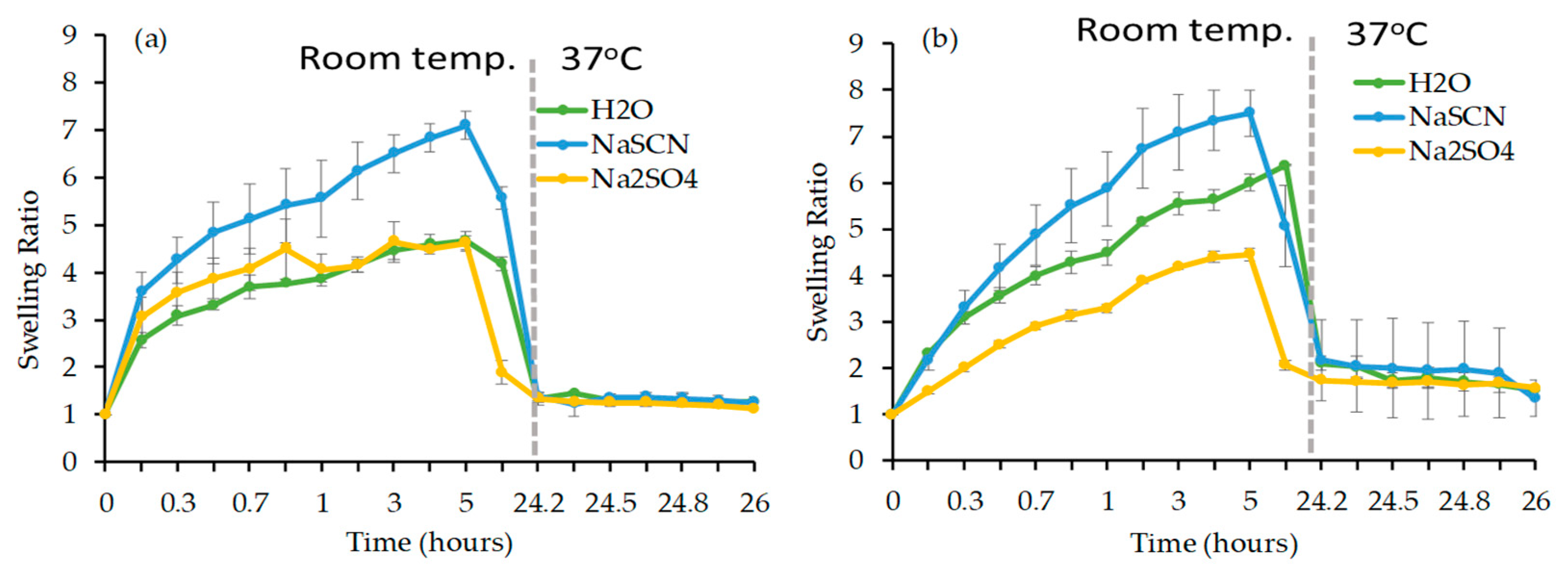
3.2.3. Evaluation of Physical Properties
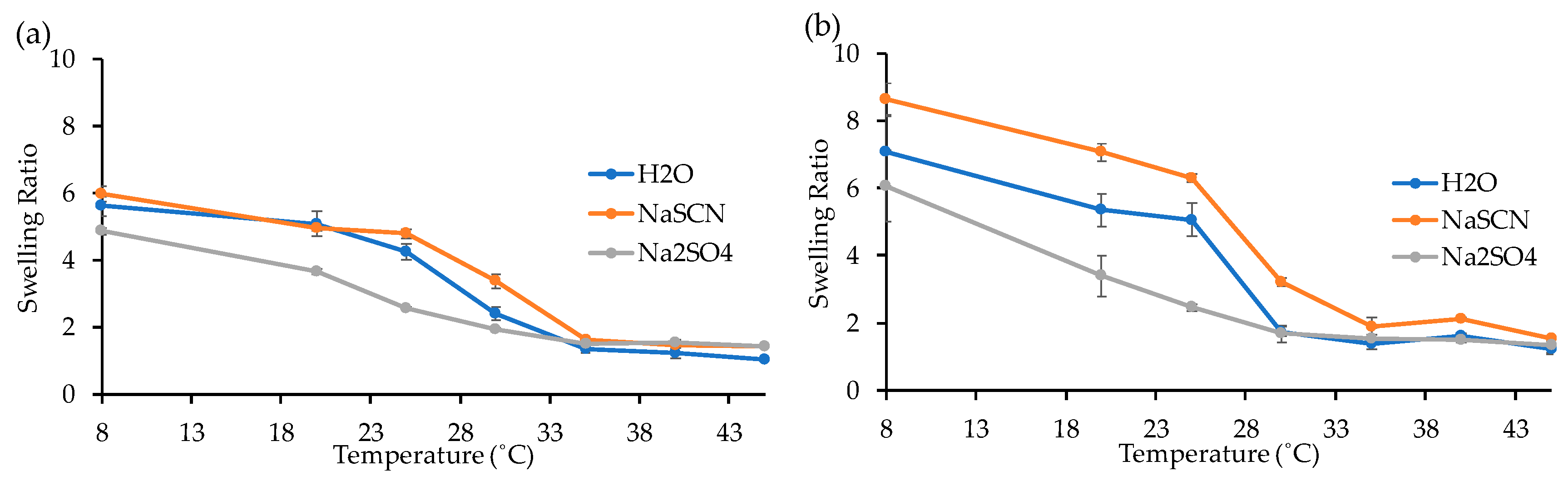
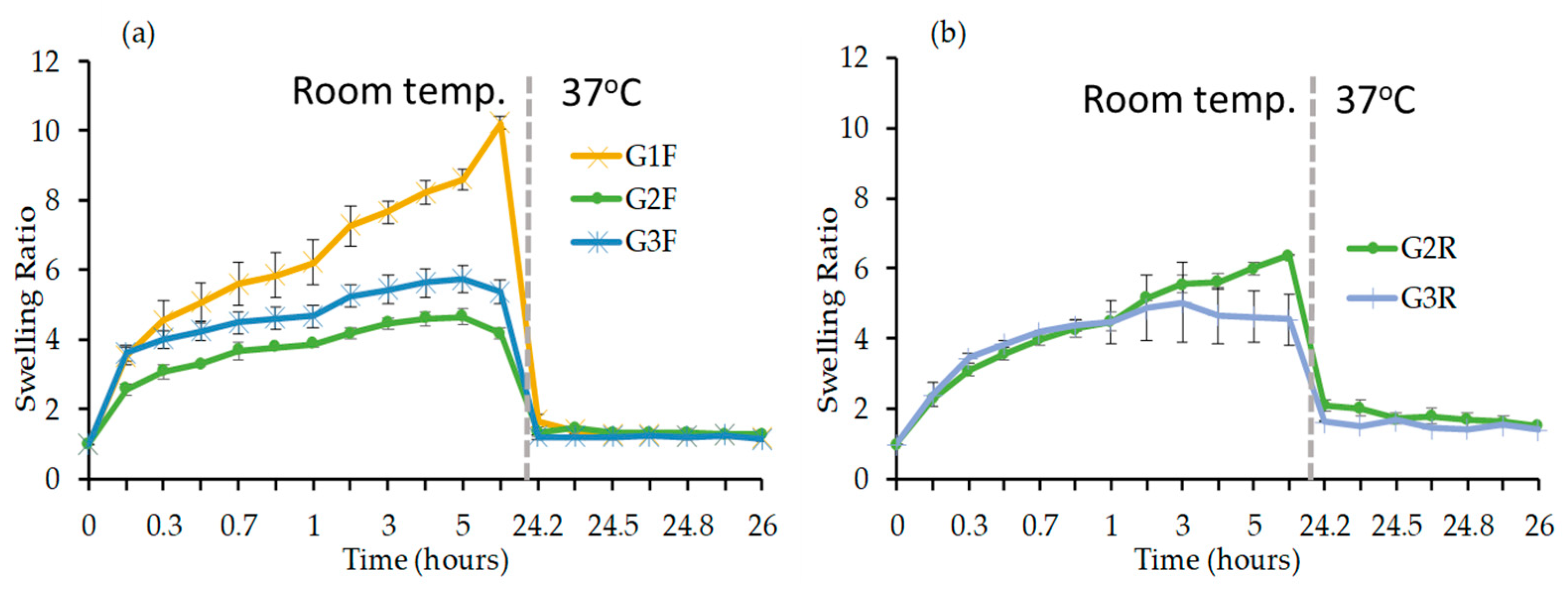
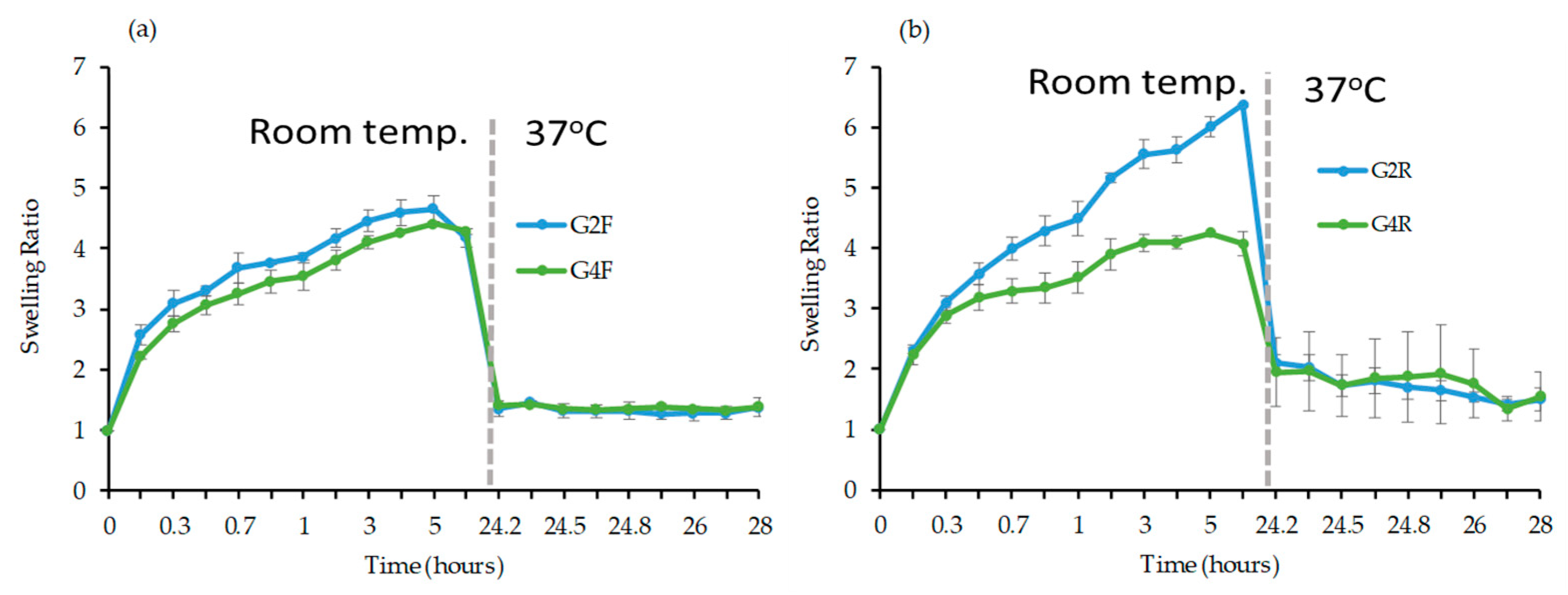
Swelling Study
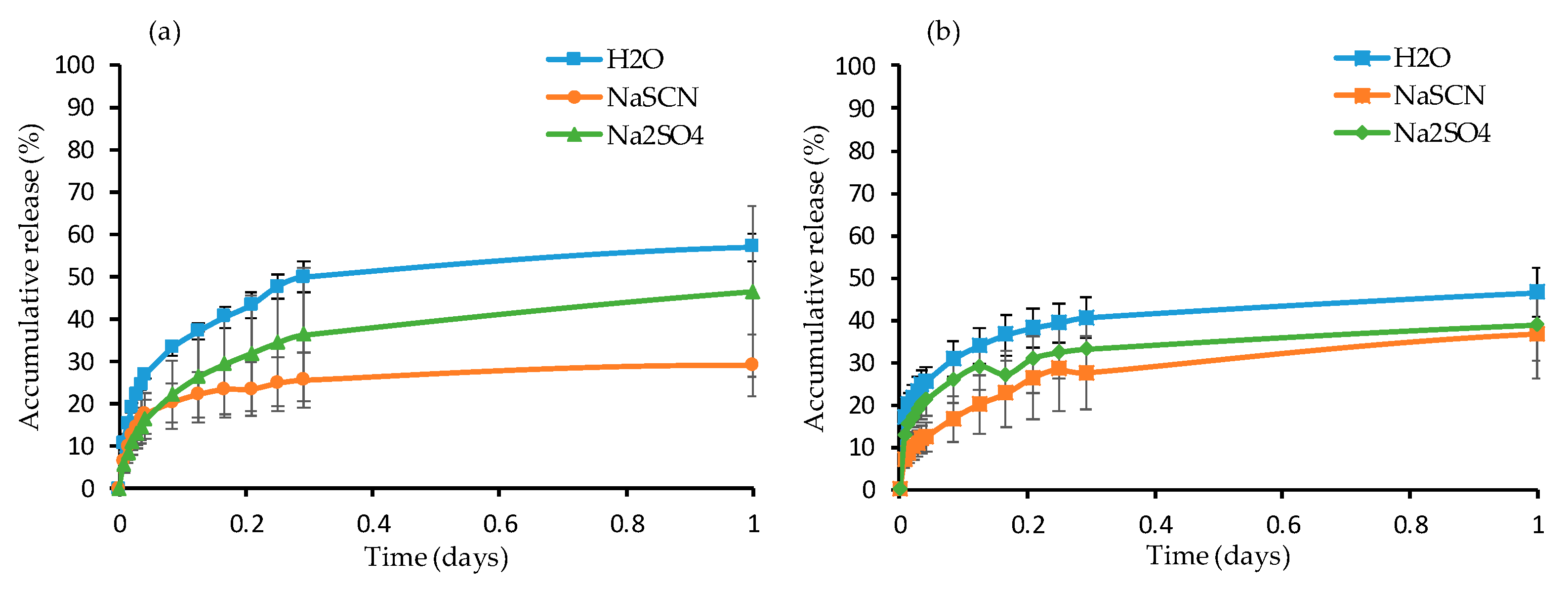
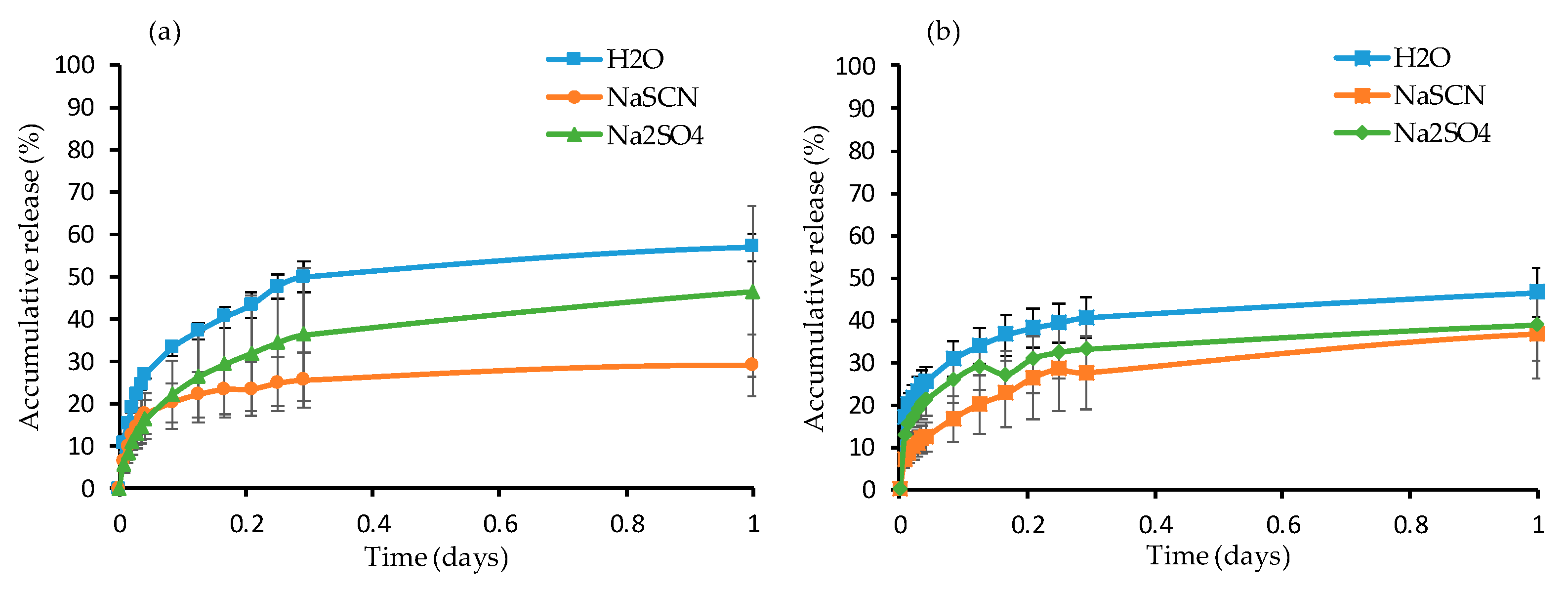
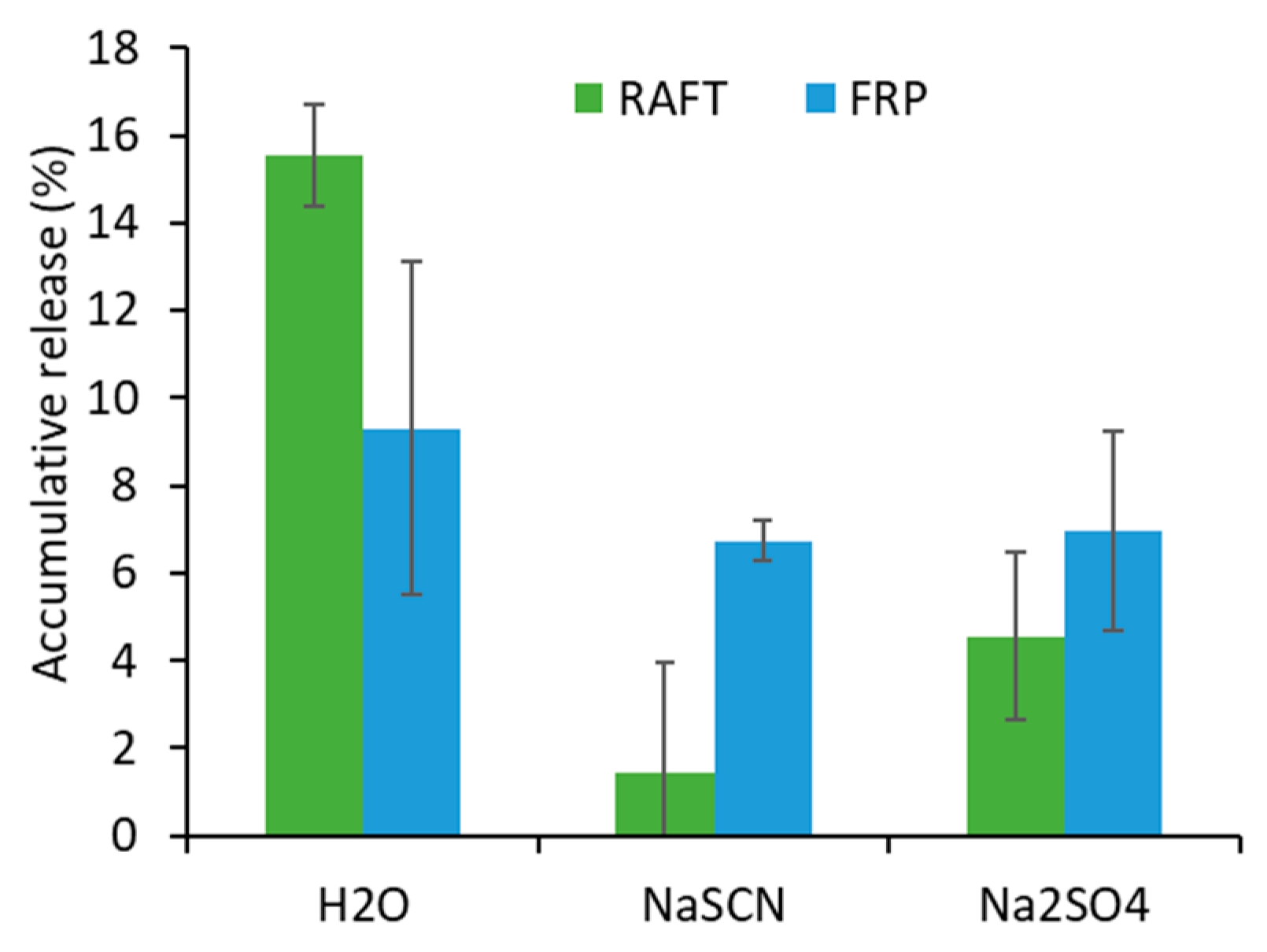
Dye Release Study
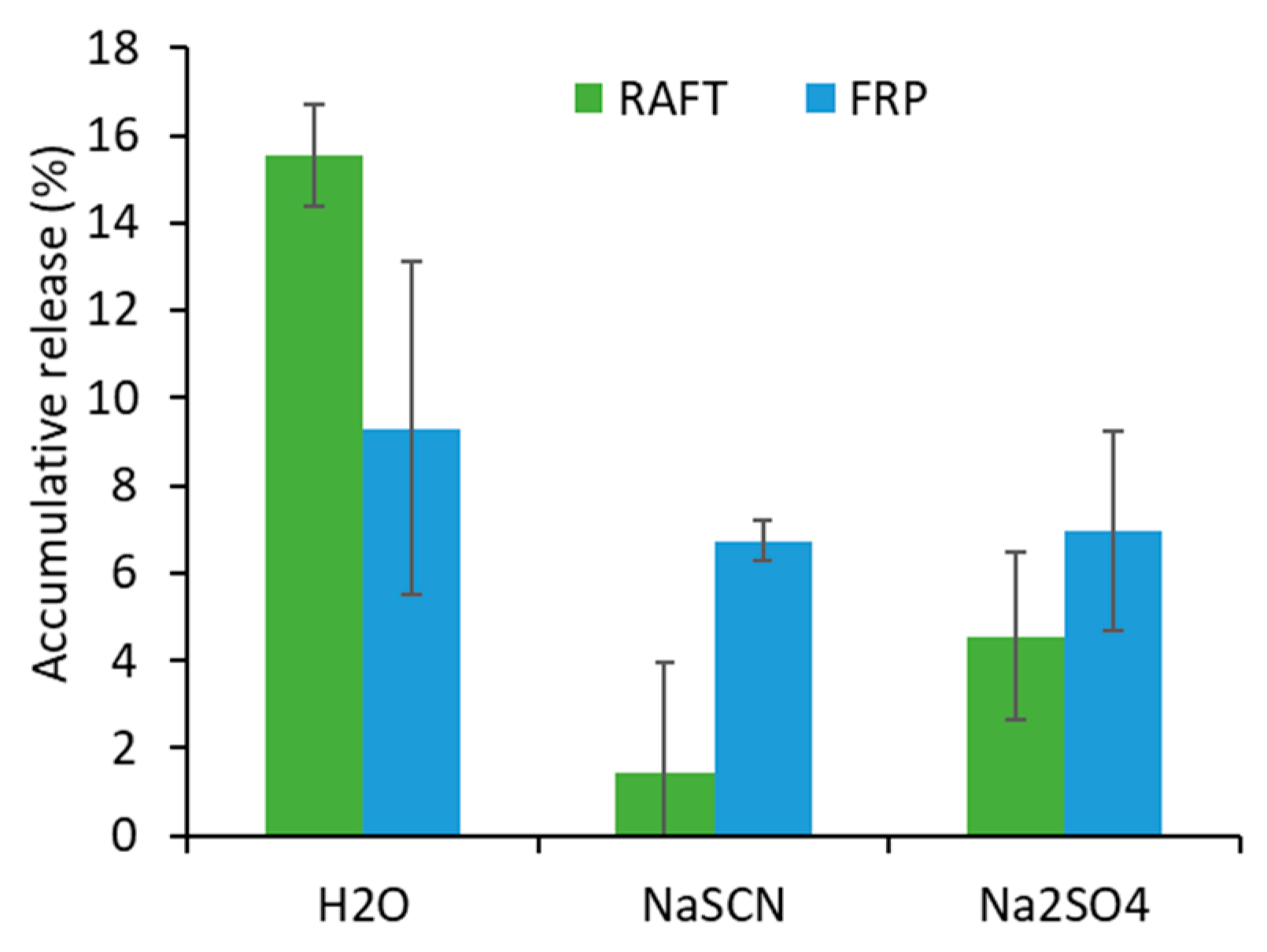
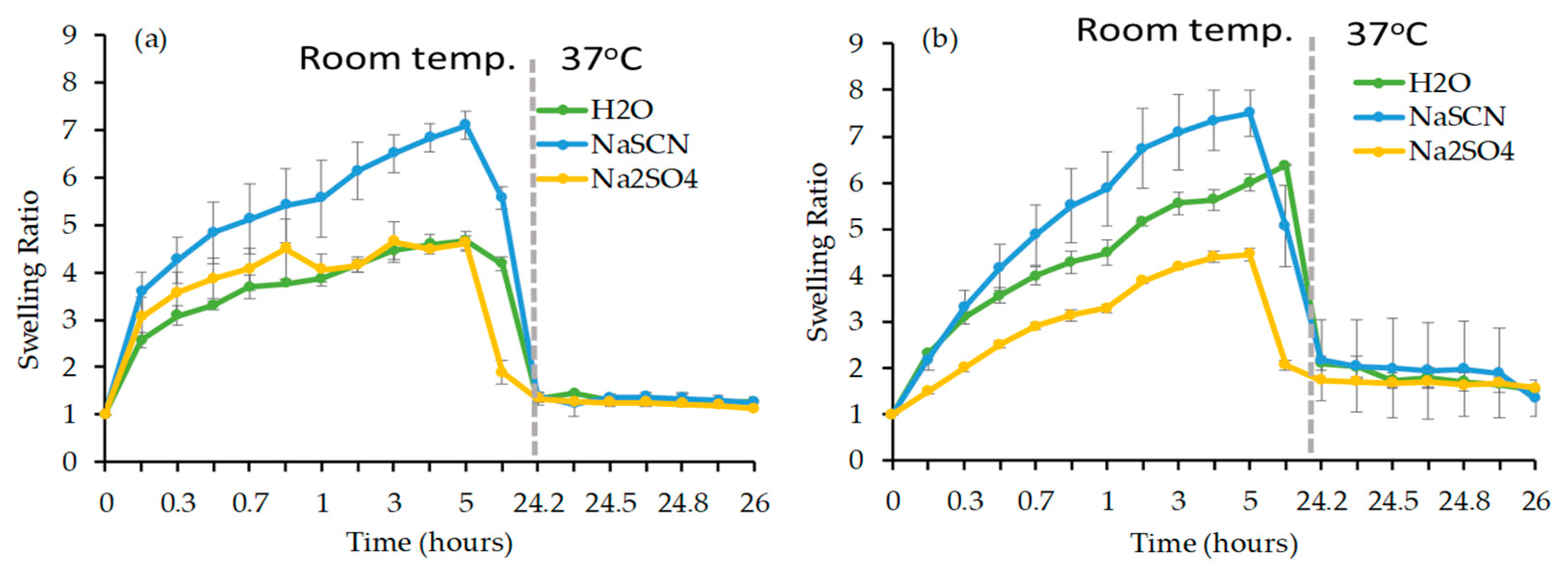
Hydrolytic Stability of Co-Monomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fairbanks, B.D.; Gunatillake, P.A.; Meagher, L. Biomedical applications of polymers derived by reversible addition–fragmentation chain-transfer (raft). Adv. Drug Deliv. Rev. 2015, 91, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Lecommandoux, S.; Garanger, É. Precision polymers with biological activity: Design towards self-assembly and bioactivity. C. R. Chim. 2016, 19, 143–147. [Google Scholar] [CrossRef]
- Pasparakis, G.; Vamvakaki, M. Multiresponsive polymers: Nano-sized assemblies, stimuli-sensitive gels and smart surfaces. Polym. Chem. 2011, 2, 1234–1248. [Google Scholar] [CrossRef]
- Ide, N.; Fukuda, T. Nitroxide-controlled free-radical copolymerization of vinyl and divinyl monomers. Evaluation of pendant-vinyl reactivity. Macromolecules 1997, 30, 4268–4271. [Google Scholar] [CrossRef]
- Ide, N.; Fukuda, T. Nitroxide-controlled free-radical copolymerization of vinyl and divinyl monomers. 2. Gelation. Macromolecules 1999, 32, 95–99. [Google Scholar] [CrossRef]
- Grubbs, R.B. Nitroxide-mediated radical polymerization: Limitations and versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar] [CrossRef]
- Gao, H.; Li, W.; Matyjaszewski, K. Synthesis of polyacrylate networks by atrp: Parameters influencing experimental gel points. Macromolecules 2008, 41, 2335–2340. [Google Scholar] [CrossRef]
- Gao, H.; Min, K.; Matyjaszewski, K. Determination of gel point during atom transfer radical copolymerization with cross-linker. Macromolecules 2007, 40, 7763–7770. [Google Scholar] [CrossRef]
- Isaure, F.; Cormack, P.A.G.; Graham, S.; Sherrington, D.C.; Armes, S.P.; Butun, V. Synthesis of branched poly (methyl methacrylate) s via controlled/living polymerisations exploiting ethylene glycol dimethacrylate as branching agent. Chem. Commun. 2004, 9, 1138–1139. [Google Scholar] [CrossRef]
- Chengfa, J.; Youqing, S.; Shiping, Z.; David, H. Gel formation in atom transfer radical polymerization of 2-(n,n-dimethylamino) ethyl methacrylate and ethylene glycol dimethacrylate. J. Polym. Sci. Part A 2001, 39, 3780–3788. [Google Scholar]
- Wang Aileen, R.; Zhu, S. Branching and gelation in atom transfer radical polymerization of methyl methacrylate and ethylene glycol dimethacrylate. Polym. Eng. Sci. 2005, 45, 720–727. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom transfer radical polymerization (atrp): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef]
- Norisuye, T.; Morinaga, T.; Tran-Cong-Miyata, Q.; Goto, A.; Fukuda, T.; Shibayama, M. Comparison of the gelation dynamics for polystyrenes prepared by conventional and living radical polymerizations: A time-resolved dynamic light scattering study. Polymer 2005, 46, 1982–1994. [Google Scholar] [CrossRef]
- Vittorio, C.; Mariella, D.; Debora, B.; Giancarlo, M. Hydrogels based on pullulan derivatives crosslinked via a “living” free-radical process. Macromol. Chem. Phys. 2002, 203, 1285–1291. [Google Scholar]
- Ding, Z.; Yingbo, R.; Xuemei, Z.; Rong, R. Kinetics of uv-initiated raft crosslinking polymerization of dimethacrylates. J. Appl. Polym. Sci. 2011, 121, 660–665. [Google Scholar]
- Barner-Kowollik, C. Handbook of Raft Polymerization; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the raft process a second update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Moad, G. Raft (reversible addition–fragmentation chain transfer) crosslinking (co)polymerization of multi-olefinic monomers to form polymer networks. Polym. Int. 2015, 64, 15–24. [Google Scholar] [CrossRef]
- Perrier, S. 50th anniversary perspective: Raft polymerization—A user guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Pérez-Salinas, P.; Jaramillo-Soto, G.; Rosas-Aburto, A.; Vázquez-Torres, H.; Bernad-Bernad, M.; Licea-Claverie, Á.; Vivaldo-Lima, E. Comparison of polymer networks synthesized by conventional free radical and raft copolymerization processes in supercritical carbon dioxide. Processes 2017, 5, 26. [Google Scholar] [CrossRef]
- Ercole, F.; Thissen, H.; Tsang, K.; Evans, R.A.; Forsythe, J.S. Photodegradable hydrogels made via raft. Macromolecules 2012, 45, 8387–8400. [Google Scholar] [CrossRef]
- Scherf, R.; Müller, L.S.; Grosch, D.; Hübner, E.G.; Oppermann, W. Investigation on the homogeneity of pmma gels synthesized via raft polymerization. Polymer 2015, 58, 36–42. [Google Scholar] [CrossRef]
- Gonçalves, M.A.D.; Pinto, V.D.; Costa, R.A.S.; Dias, R.C.S.; Hernándes-Ortiz, J.C.; Costa, M.R.P.F.N. Stimuli-responsive hydrogels synthesis using free radical and raft polymerization. Macromol. Symp. 2013, 333, 41–54. [Google Scholar] [CrossRef]
- Yoon, J.A.; Gayathri, C.; Gil, R.R.; Kowalewski, T.; Matyjaszewski, K. Comparison of the thermoresponsive deswelling kinetics of poly(2-(2-methoxyethoxy) ethyl methacrylate) hydrogels prepared by atrp and frp. Macromolecules 2010, 43, 4791–4797. [Google Scholar] [CrossRef]
- Gonzato, C.; Pasetto, P.; Bedoui, F.; Mazeran, P.-E.; Haupt, K. On the effect of using raft and frp for the bulk synthesis of acrylic and methacrylic molecularly imprinted polymers. Polym. Chem. 2014, 5, 1313–1322. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive polymers for biomedical applications. Polymers 2011, 3, 1215–1242. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, Y.; Huo, Y. Thermo-responsive hydrogels with n-isopropylacrylamide/acrylamide interpenetrating networks for controlled drug release. J. Biomater. Sci. Polym. Ed. 2015, 26, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Lanzalaco, S.; Armelin, E. Poly(n-isopropylacrylamide) and copolymers: A review on recent progresses in biomedical applications. Gels 2017, 3, 36. [Google Scholar] [CrossRef]
- Zhang, Y.; Furyk, S.; Bergbreiter, D.E.; Cremer, P.S. Specific ion effects on the water solubility of macromolecules: Pnipam and the hofmeister series. J. Am. Chem. Soc. 2005, 127, 14505–14510. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(n-isopropylacrylamide). J. Macromol. Sci. A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(n-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Furyk, S.; Sagle, L.B.; Cho, Y.; Bergbreiter, D.E.; Cremer, P.S. Effects of hofmeister anions on the lcst of pnipam as a function of molecular weight. J. Phys. Chem. C 2007, 111, 8916–8924. [Google Scholar] [CrossRef] [PubMed]
- De Rybel, N.; Van Steenberge, P.H.M.; Reyniers, M.F.; Barner-Kowollik, C.; D’hooge, D.R.; Marin, G.B. An update on the pivotal role of kinetic modeling for the mechanistic understanding and design of bulk and solution raft polymerization. Macromol. Theory Simul. 2017, 26, 1600048. [Google Scholar] [CrossRef]
- Wang, A.R.; Zhu, S. Effects of diffusion-controlled radical reactions on raft polymerization. Macromol. Theory Simul. 2003, 12, 196–208. [Google Scholar] [CrossRef]
- Favier, A.; Charreyre, M.T. Experimental requirements for an efficient control of free-radical polymerizations via the reversible addition-fragmentation chain transfer (raft) process. Macromol. Rapid Commun. 2006, 27, 653–692. [Google Scholar] [CrossRef]
- Yu, Q.; Zhu, Y.; Ding, Y.; Zhu, S. Reaction behavior and network development in raft radical polymerization of dimethacrylates. Macromol. Chem. Phys. 2008, 209, 551–556. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gel_ID | Crosslinker | Vdioxane (mL) | Monomer Concentration (M) | Gel Fraction (%) | |||
|---|---|---|---|---|---|---|---|
| mg | mmol | %mol | FRP | RAFT | |||
| G1 | 7.2 | 0.047 | 2.6 | 1 | 1.814 | 92.0 | – |
| G2 | 14.3 | 0.093 | 5 | 1 | 2.697 | 79.2 | 76.3 |
| G3 | 28.7 | 0.186 | 10 | 1 | 1.953 | 79.2 | 76.3 |
| G4 | 14.3 | 0.093 | 5 | 0.5 | 3.720 | 82.9 | 88.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joubert, F.; Cheong Phey Denn, P.; Guo, Y.; Pasparakis, G. Comparison of Thermoresponsive Hydrogels Synthesized by Conventional Free Radical and RAFT Polymerization. Materials 2019, 12, 2697. https://doi.org/10.3390/ma12172697
Joubert F, Cheong Phey Denn P, Guo Y, Pasparakis G. Comparison of Thermoresponsive Hydrogels Synthesized by Conventional Free Radical and RAFT Polymerization. Materials. 2019; 12(17):2697. https://doi.org/10.3390/ma12172697
Chicago/Turabian StyleJoubert, Fanny, Peyton Cheong Phey Denn, Yujie Guo, and George Pasparakis. 2019. "Comparison of Thermoresponsive Hydrogels Synthesized by Conventional Free Radical and RAFT Polymerization" Materials 12, no. 17: 2697. https://doi.org/10.3390/ma12172697
APA StyleJoubert, F., Cheong Phey Denn, P., Guo, Y., & Pasparakis, G. (2019). Comparison of Thermoresponsive Hydrogels Synthesized by Conventional Free Radical and RAFT Polymerization. Materials, 12(17), 2697. https://doi.org/10.3390/ma12172697