Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison


 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Preparation
2.2. Sample Characterization
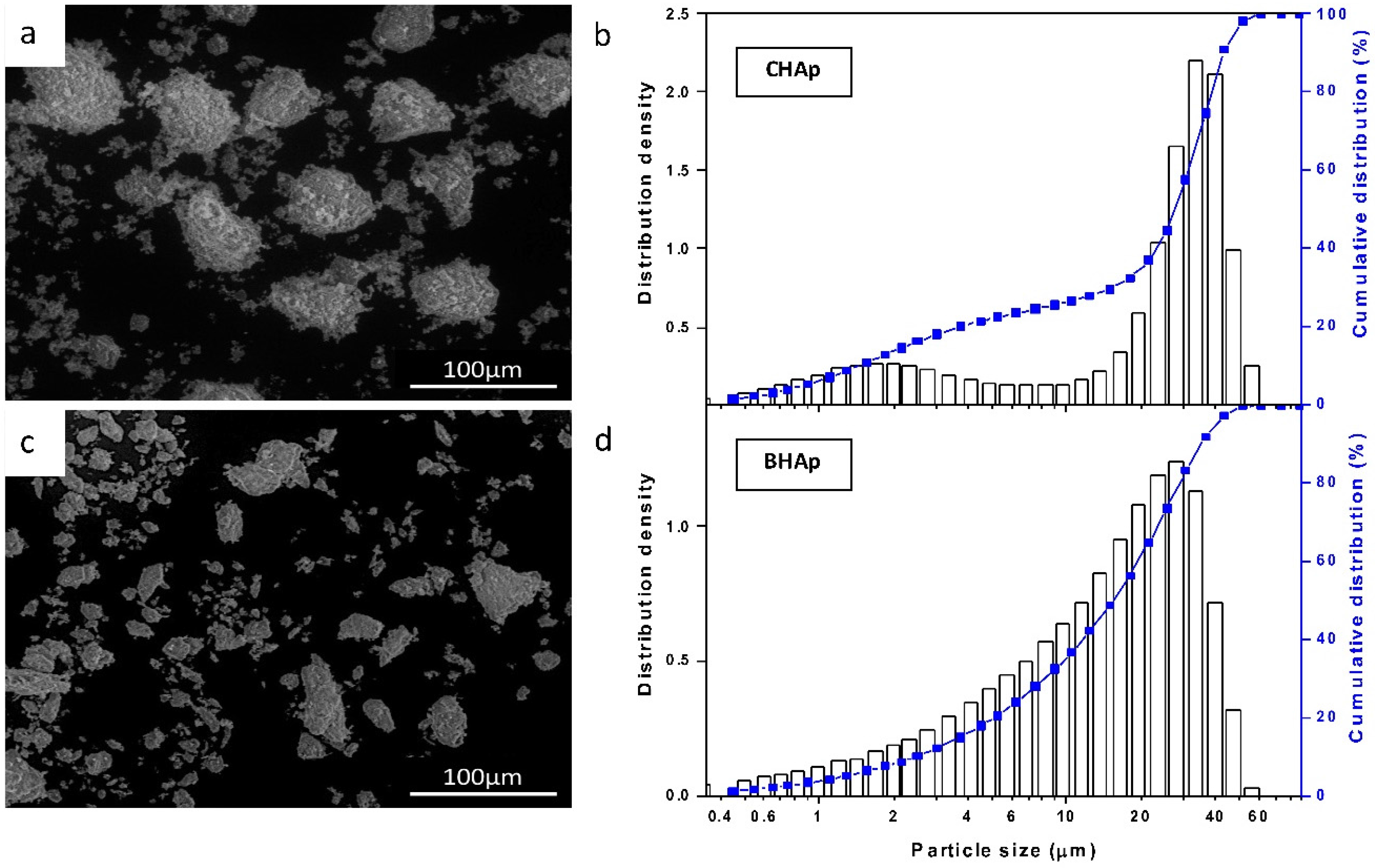
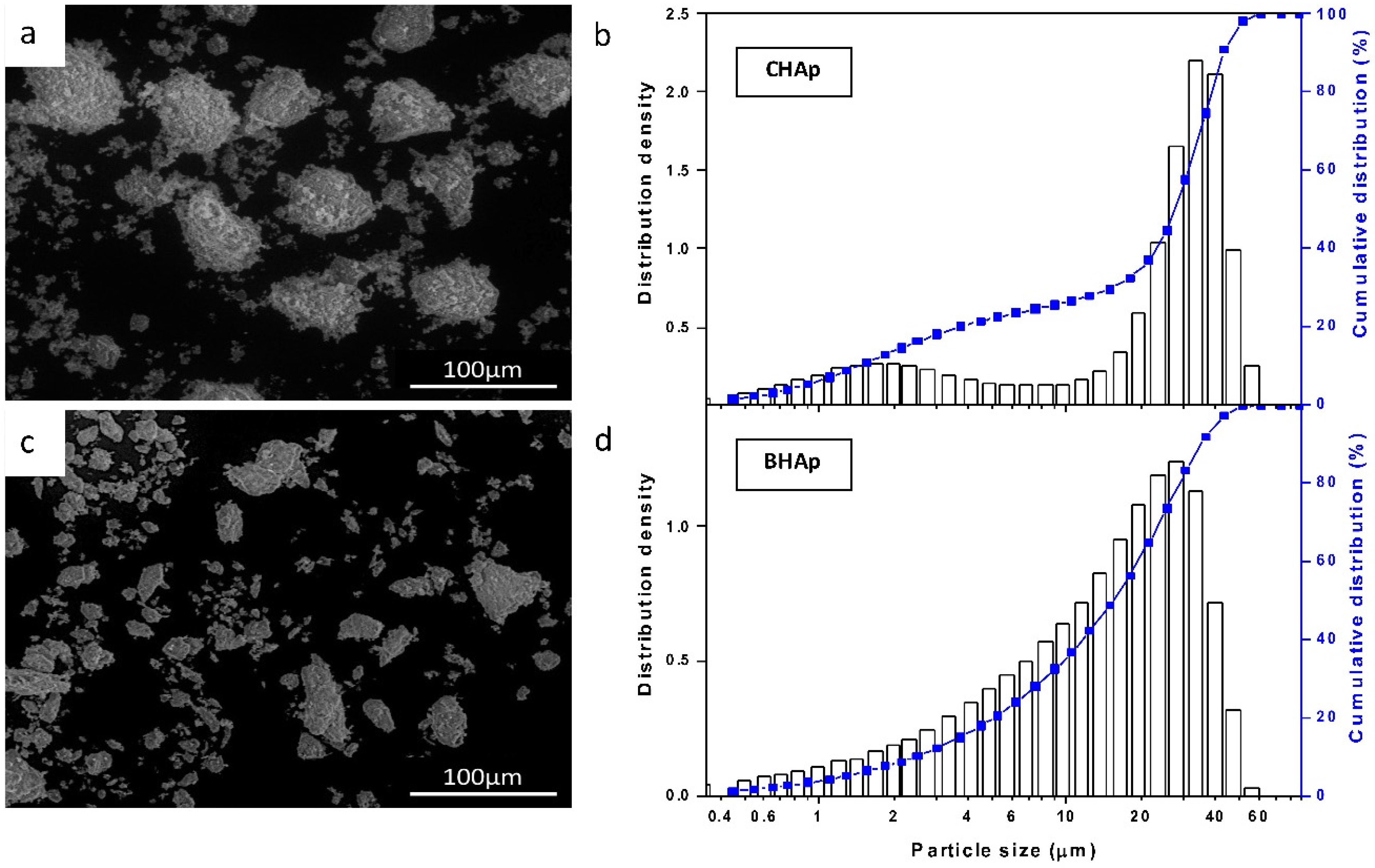
2.2.1. Powder Size Distribution, Chemical Composition and Microstructure of Ceramics
2.2.2. Mechanical Properties
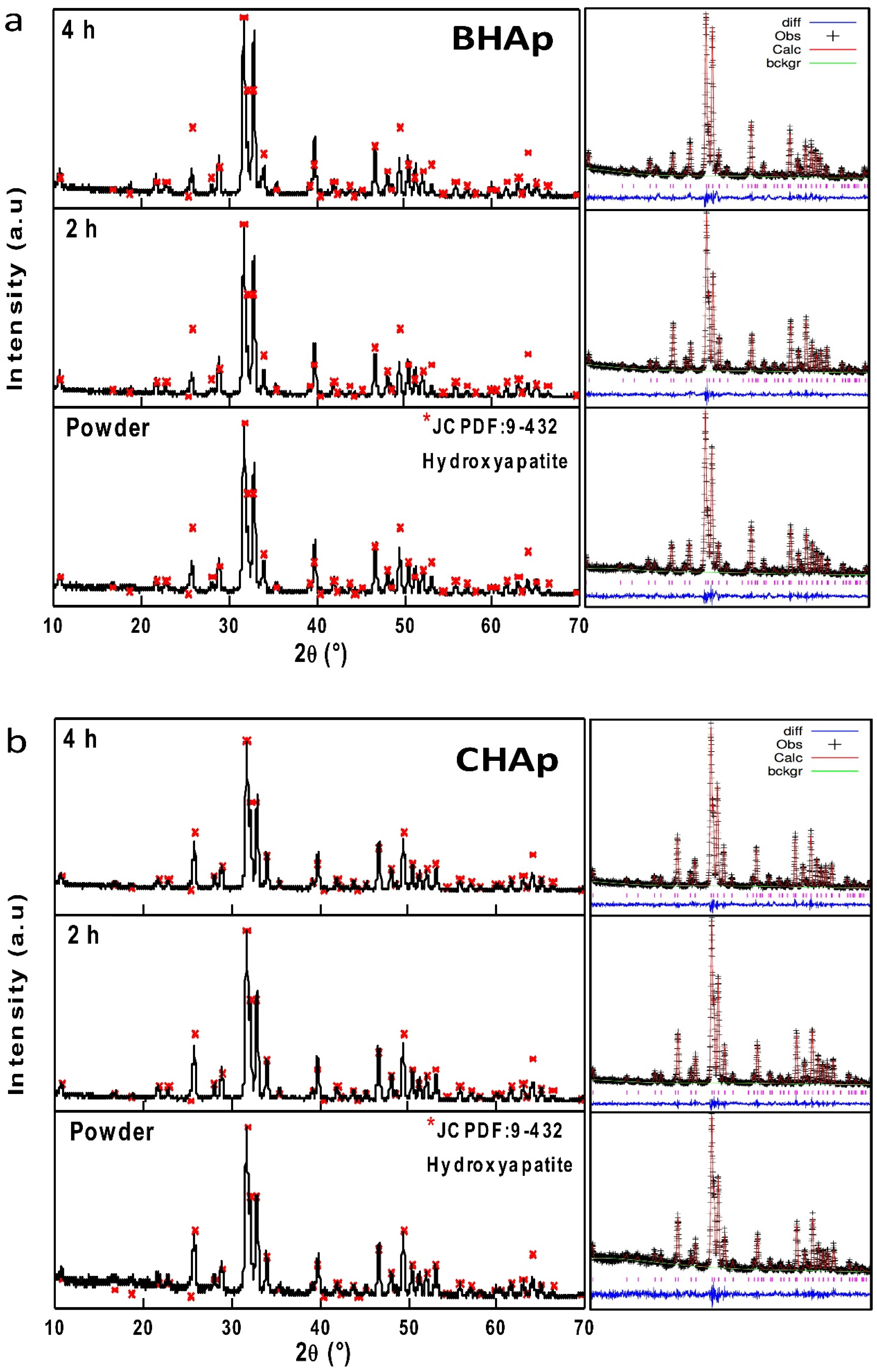
2.2.3. Structural Characteristics
2.3. Biocompatibility Assessment
2.3.1. Simulated Body Fluid (SBF) Immersion
2.3.2. Cell Culture and Exposure
- Cell mitochondrial activity
- Cytotoxicity determination
- Statistical analysis
3. Results and Discussion
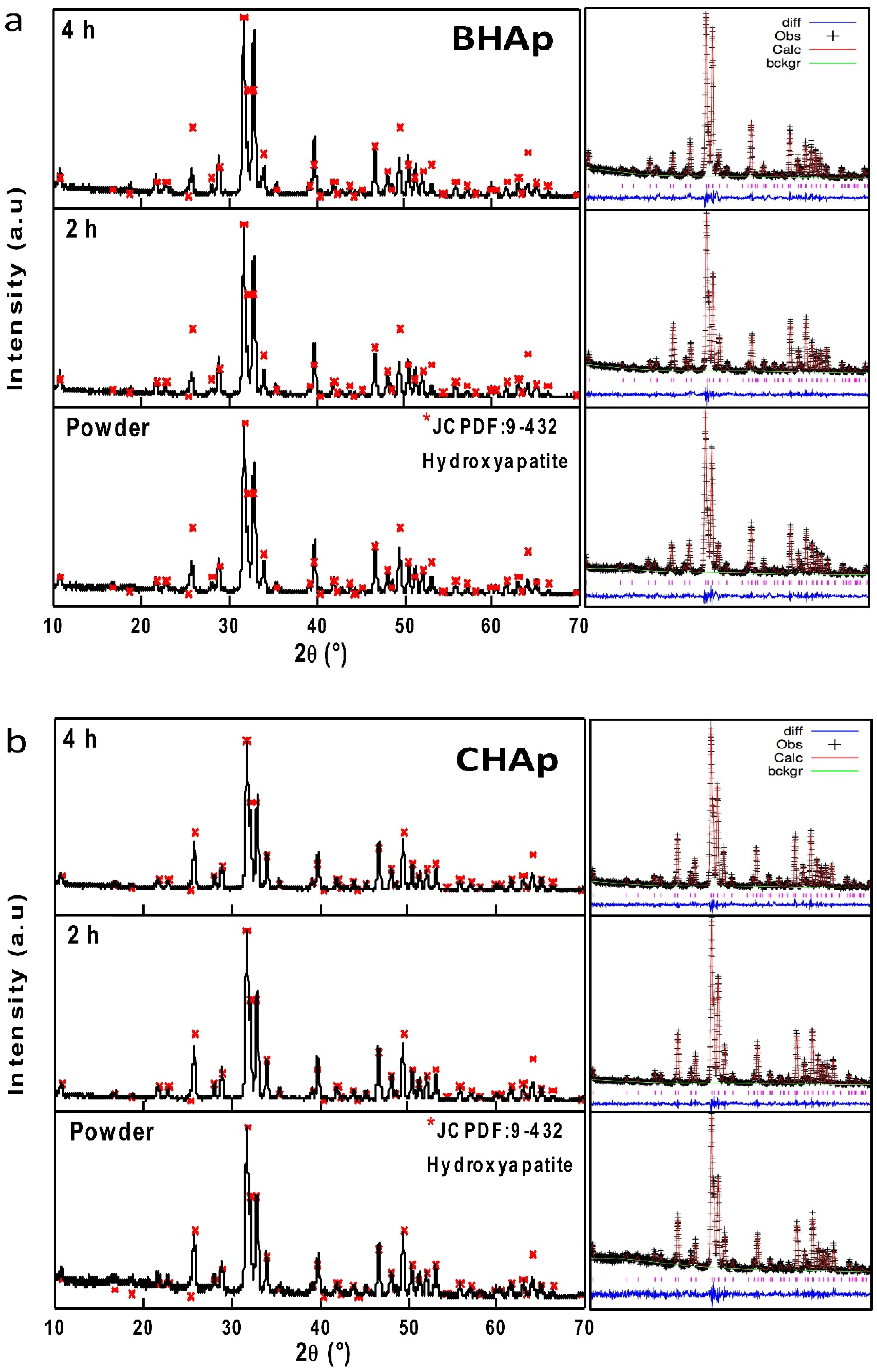
3.1. Microstructure
3.2. Structural Analysis
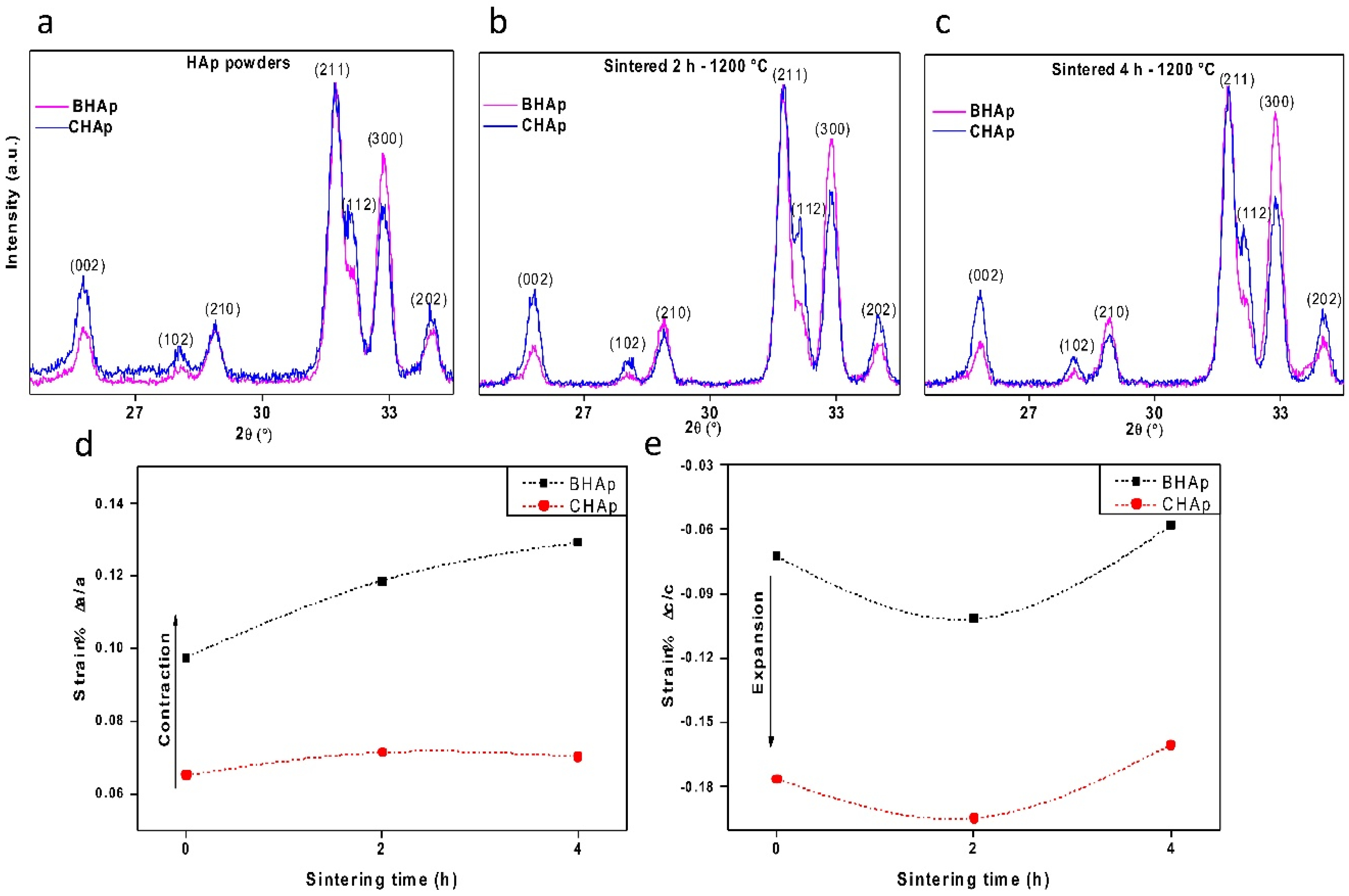
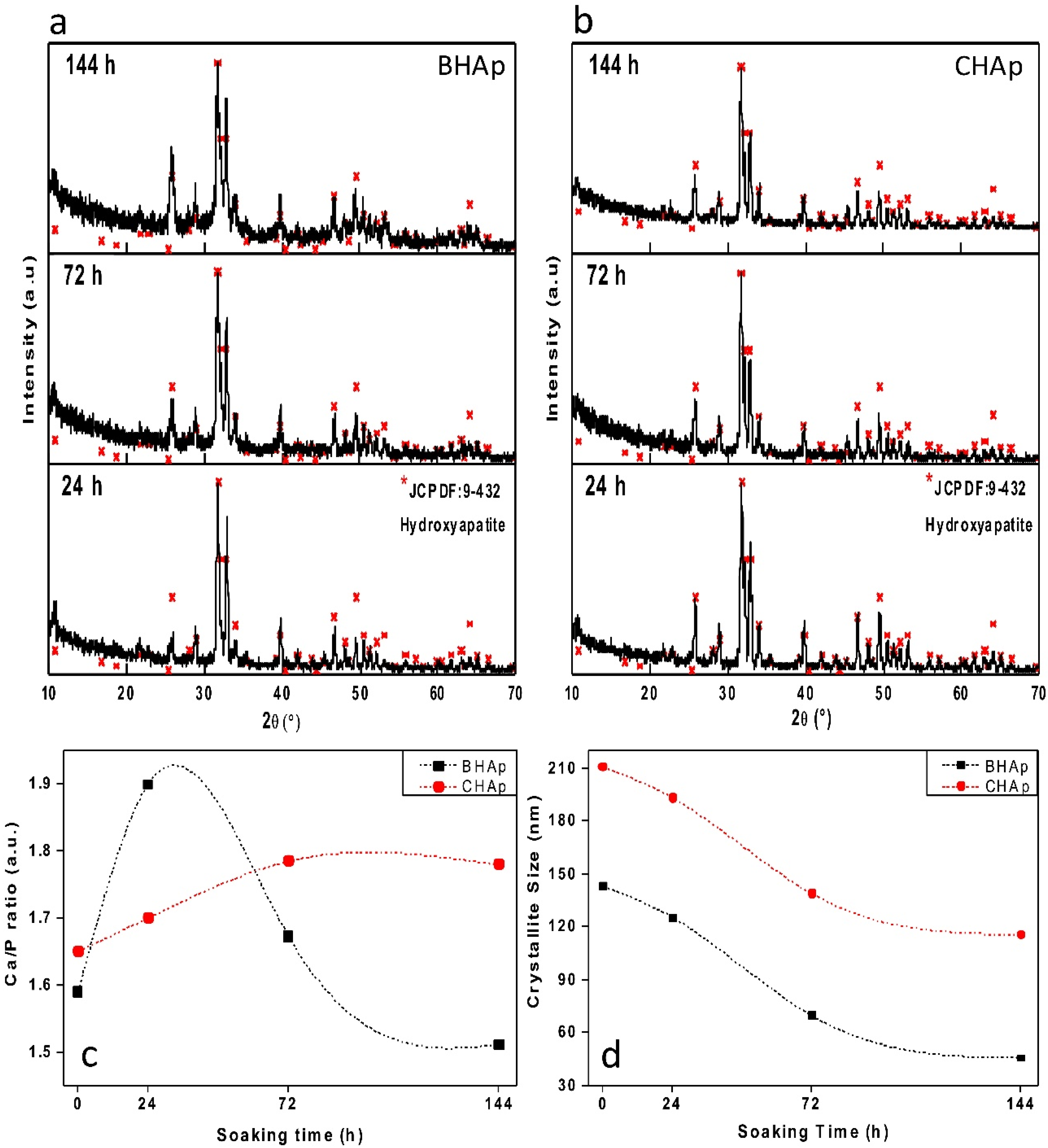
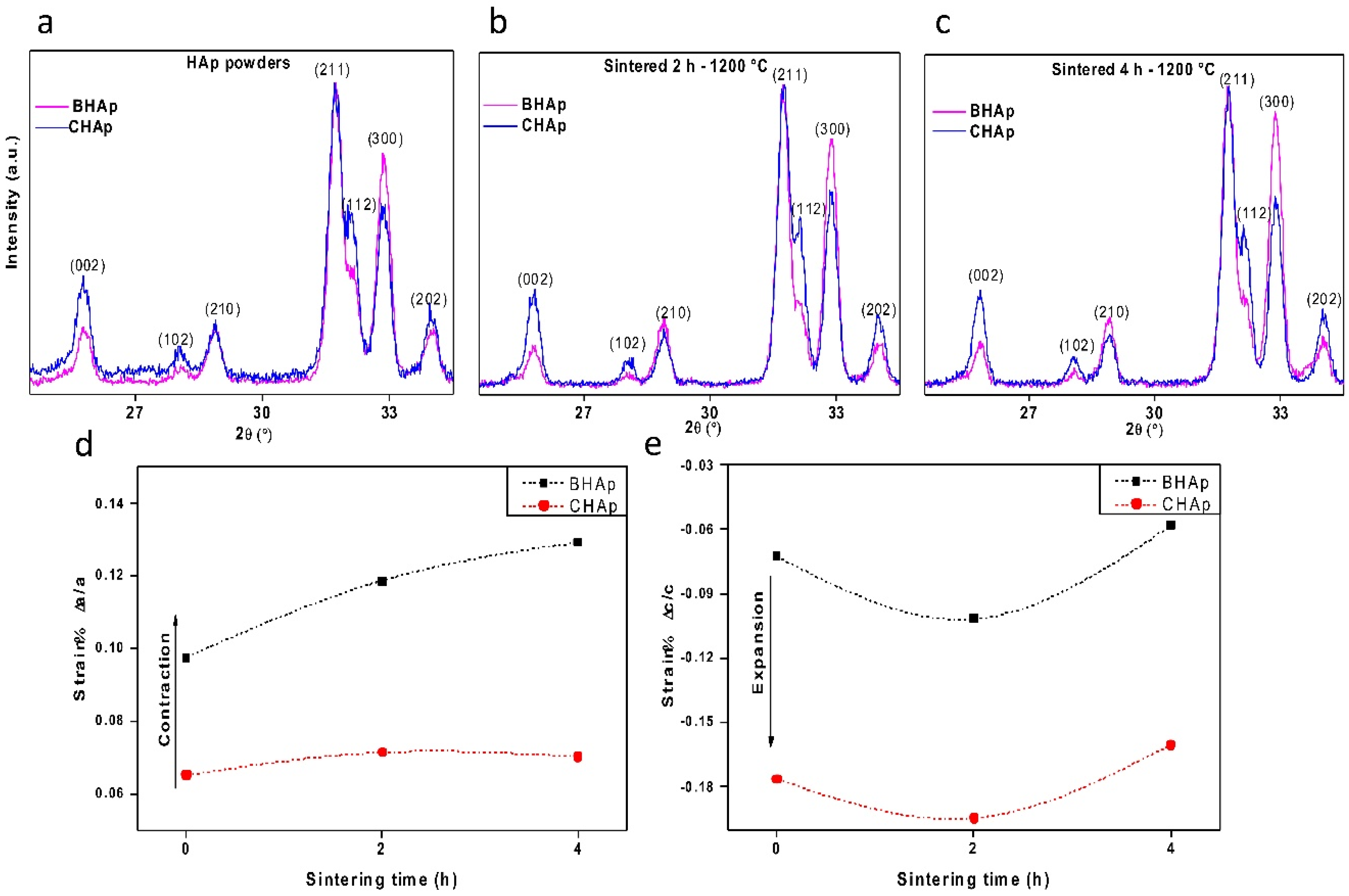
3.2.1. Crystallographic Details (XRD)
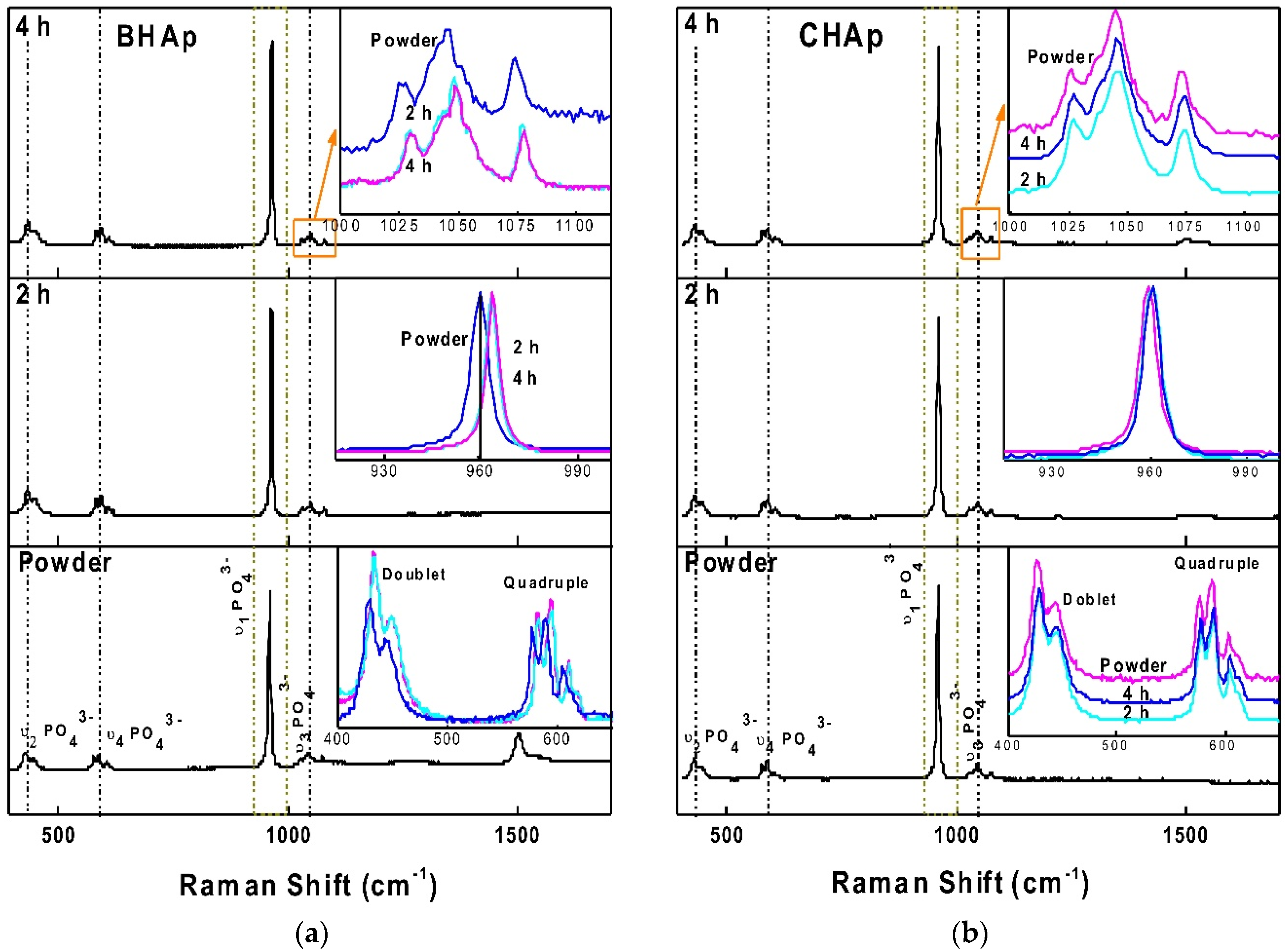
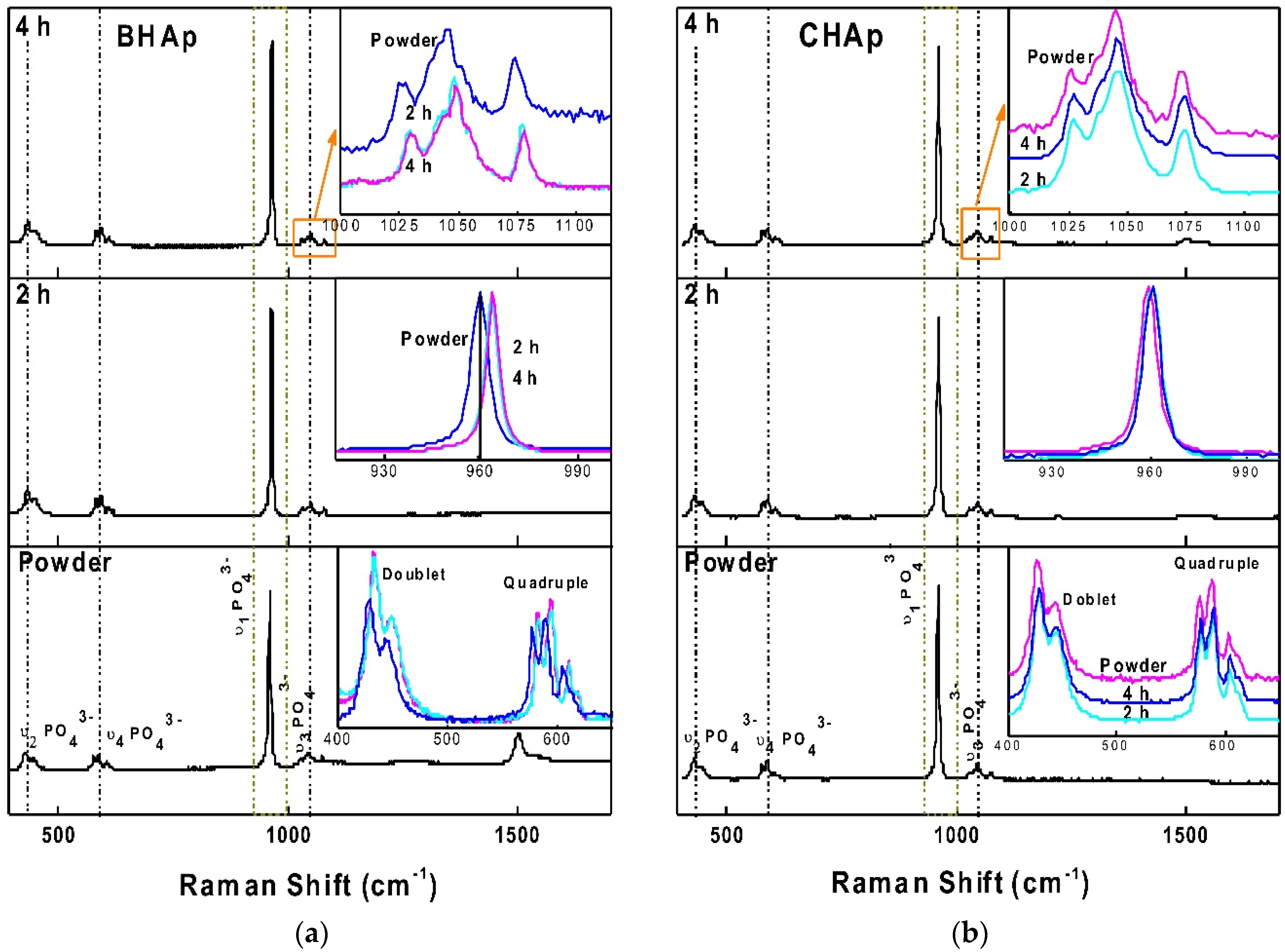
3.2.2. Raman Spectroscopy
3.3. In Vitro Assessment in SBF
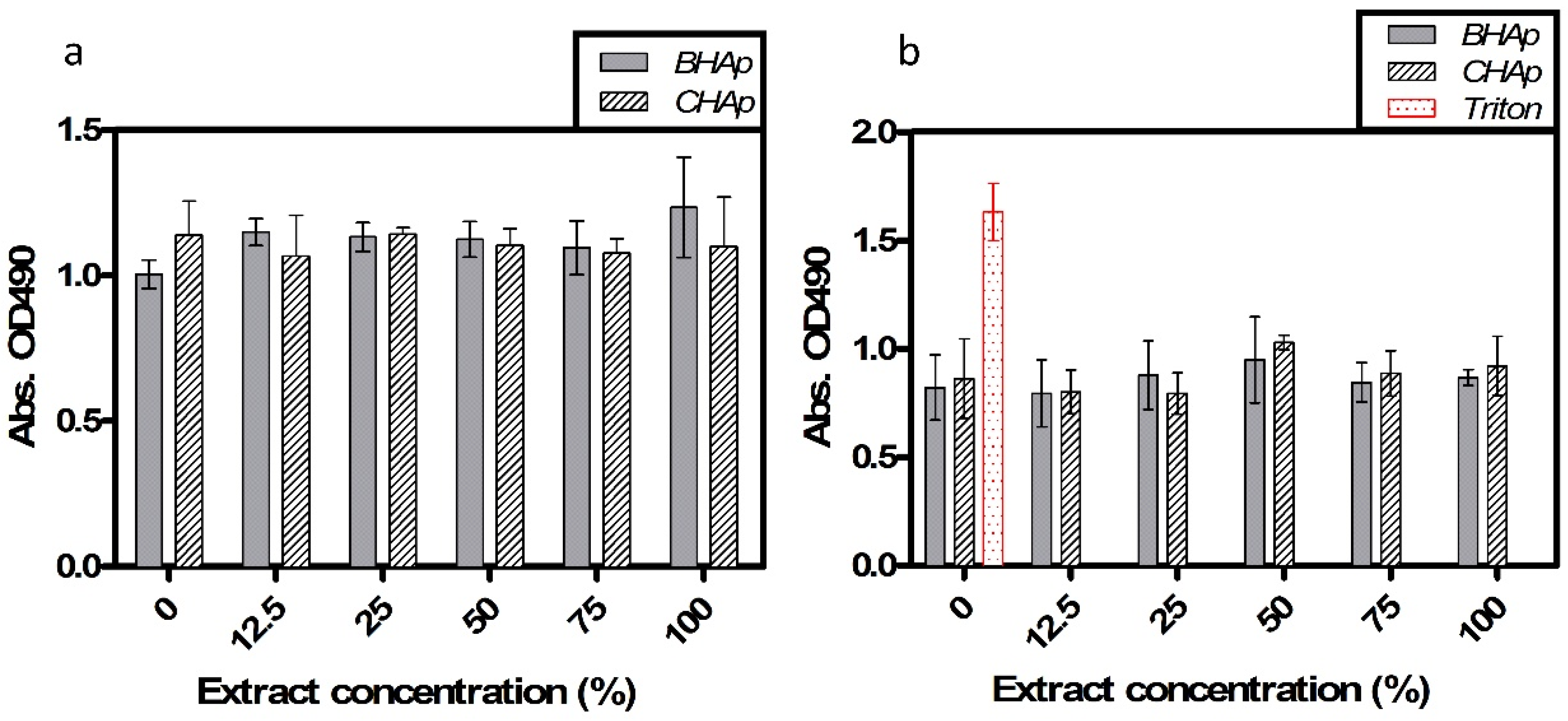
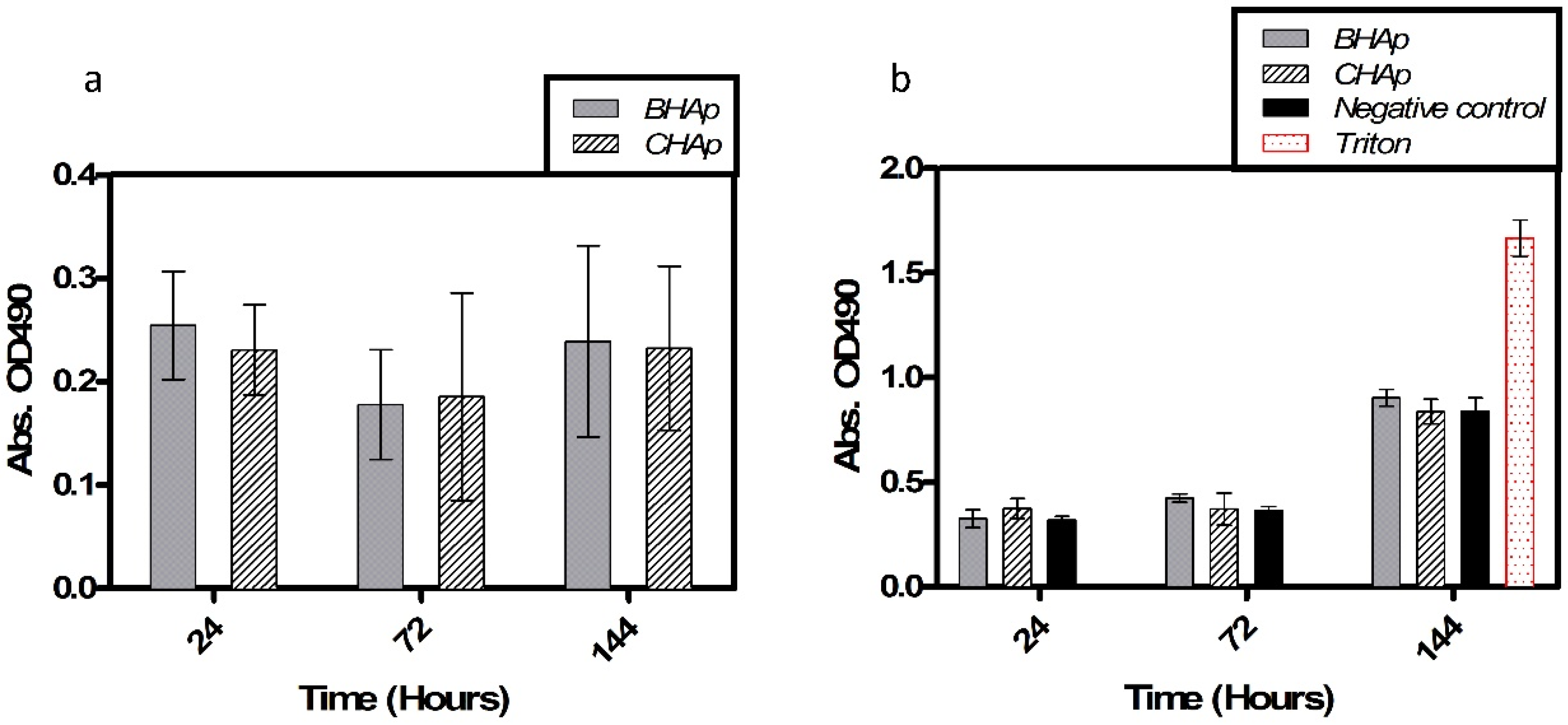
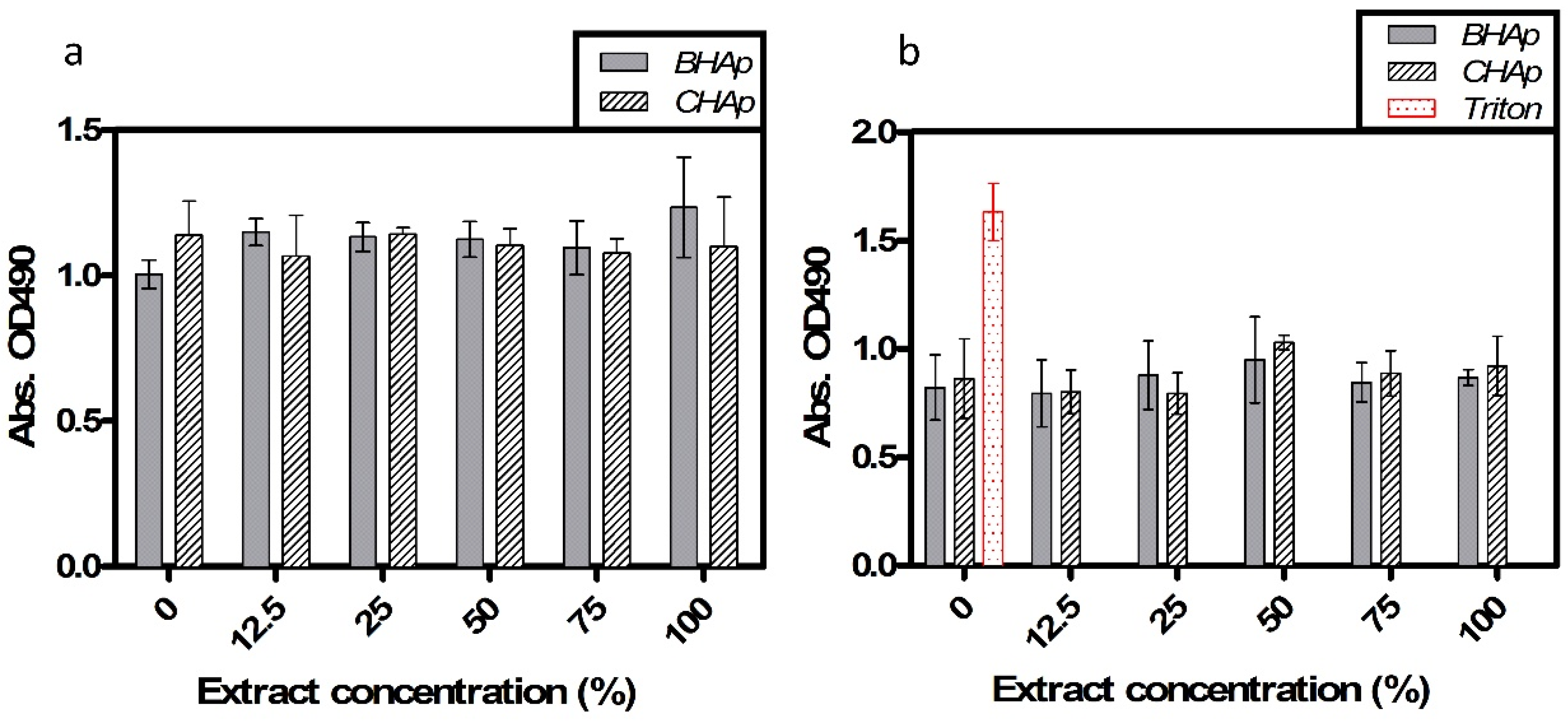
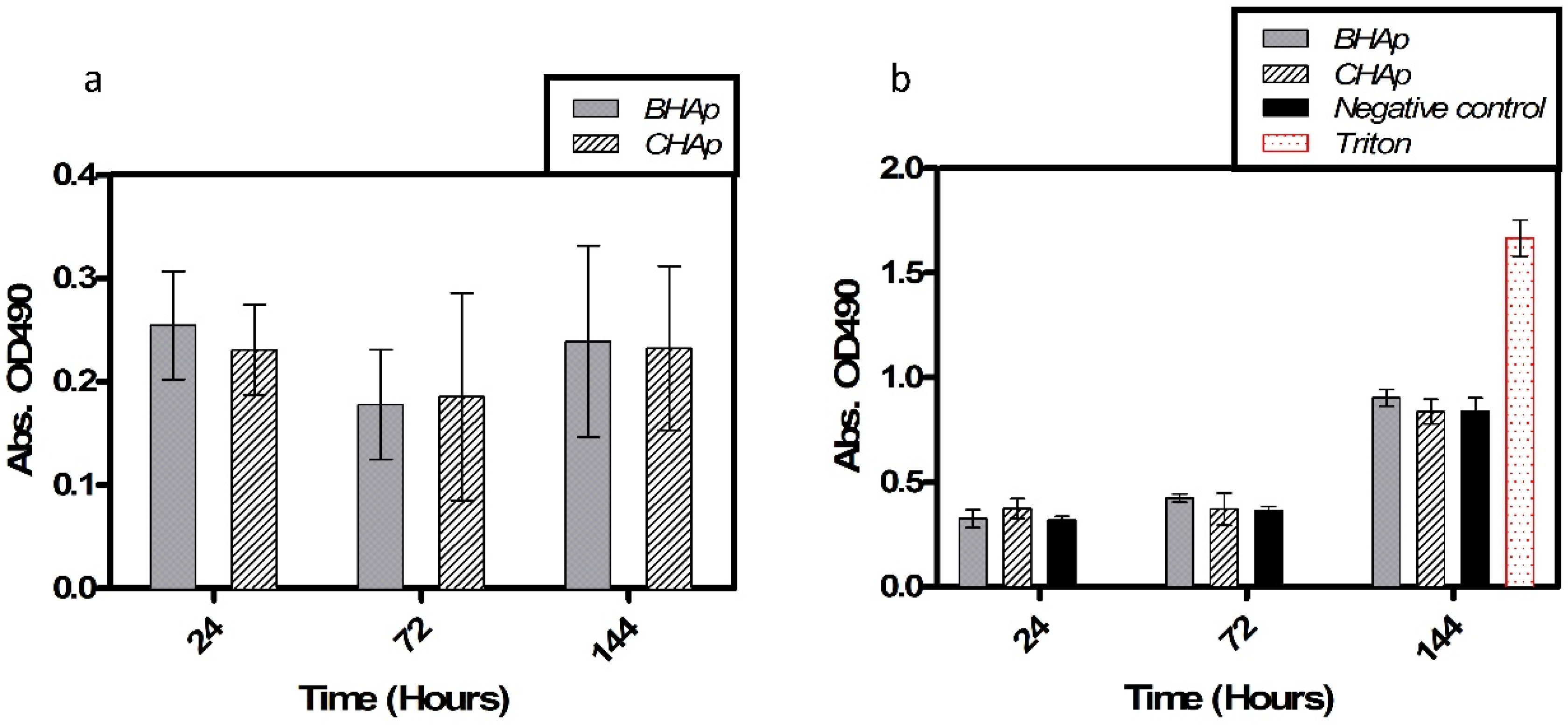
3.4. In Vitro Assessment in Cell Culture
4. Conclusions
- The in-house developed processing route allowed obtaining highly crystalline HAp from bovine bone. The Na+ and Mg2+ ions intrinsically presented in BHAp seem to influence the sintering behavior evolving to ceramics with lower porosity and coarser microstructure compared to those obtained with synthetic HAp.
- At the structural level, the main differences between the BHAp and CHAp consist in an increase in the orientation degree of a,b-plane and the contraction of a lattice parameter of HAp unit cell. These differences are related to the BHAp source which may have a positive effect in the in vitro performance, quantifiable at longer immersion periods.
- Despite the clear structural differences between BHAp and CHAp at the unit cell level, the hydroxyapatite obtained from bovine bones has comparable in vitro behavior with the commercial one. This result is very significant considering the positive cost/benefit ratio from BHAp and thus can be used with no restrictions for biomedical applications in the same way as CHAp.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poitout, D.G. Biomechanics and Biomaterials in Orthopedics; Springer: London, UK, 2004; ISBN 978-1-4471-3774-0. [Google Scholar]
- Basu, B.; Ghosh, S. Biomaterials for Musculoskeletal Regeneration; Springer: Bangalore, India, 2017; ISBN 9789811030581. [Google Scholar]
- Kokubo, T. Bioactive glass ceramics: properties and applications. Biomaterials 1991, 12, 155–163. [Google Scholar] [CrossRef]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Lin, K.; Zhou, Y.; Zhou, Y.; Qu, H.; Chen, F.; Zhu, Y.; Chang, J. Biomimetic hydroxyapatite porous microspheres with co-substituted essential trace elements: Surfactant-free hydrothermal synthesis, enhanced degradation and drug release. J. Mater. Chem. 2011, 21, 16558. [Google Scholar] [CrossRef]
- Wiglusz, R.J.; Kedziora, A.; Lukowiak, A.; Doroszkiewicz, W.; Strek, W. Hydroxyapatites and Europium(III) doped hydroxyapatites as a carrier of silver nanoparticles and their antimicrobial activity. J. Biomed. Nanotechnol. 2012, 8, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Huang, Y. Physical Properties of Nano-HAs/ZrO 2 Coating on Surface of Titanium Materials Used in Dental-Implants and Its Biological Compatibility. J. Nanosci. Nanotechnol. 2012, 12, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Astala, R.; Stott, M.J. First principles investigation of mineral component of bone: CO3 substitutions in hydroxyapatite. Chem. Mater. 2005, 17, 4125–4133. [Google Scholar] [CrossRef]
- Elliott, J.C.; Wilson, R.M.; Dowker, S.E.P. Apatite structures. Adv. X-ray Anal. 2002, 45, 172–181. [Google Scholar] [CrossRef]
- Sudarsanan, K.; Young, R.A. Significant precision in crystal structural details. Holly Springs hydroxyapatite. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1969, 25, 1534–1543. [Google Scholar] [CrossRef]
- Murugan, R.; Kumar, T.S.S.; Rao, K.P. Fluorinated bovine hydroxyapatite: Preparation and characterization. Mater. Lett. 2002, 57, 429–433. [Google Scholar] [CrossRef]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng. C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- Akram, M.; Ahmed, R.; Shakir, I.; Ibrahim, W.A.W.; Hussain, R. Extracting hydroxyapatite and its precursors from natural resources. J. Mater. Sci. 2014, 49, 1461–1475. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Mattioli-Belmonte, M.; Celotti, G.; Sandri, M.; Gigante, A.; Fava, P.; Biagini, G. Biomimetic Mg- and Mg,CO3-substituted hydroxyapatites: Synthesis characterization and in vitro behaviour. J. Eur. Ceram. Soc. 2006, 26, 2593–2601. [Google Scholar] [CrossRef]
- Barakat, N.A.M.; Khil, M.S.; Omran, A.M.; Sheikh, F.A.; Kim, H.Y. Extraction of pure natural hydroxyapatite from the bovine bones bio waste by three different methods. J. Mater. Process. Technol. 2009, 209, 3408–3415. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef] [PubMed]
- Giraldo-Betancur, A.L.; Espinosa-Arbelaez, D.G.; Del Real-López, A.; Millan-Malo, B.M.; Rivera-Muñoz, E.M.; Gutierrez-Cortez, E.; Pineda-Gomez, P.; Jimenez-Sandoval, S.; Rodriguez-García, M.E. Comparison of physicochemical properties of bio and commercial hydroxyapatite. Curr. Appl. Phys. 2013, 13, 1383–1390. [Google Scholar] [CrossRef]
- ASTM E112-13. Standard Test Methods for Determining Average Grain Size; American Society for Testing Materials: West Conshohocken, PA, USA, 1996. [Google Scholar]
- Larson, A.C.; Dreele, R.B.; Von; Alamos, L. General Structural Analysis System (GSAS); Los Alamos National Laboratory Report LAUR; Los Alamos National Laboratory: Los Alamos, NM, USA, 2000.
- Pérez Alcázar, G.A.; Colorado Restrepo, H.D. Difracción de Rayos X y el Método Rietveld Teoría y Software de Refinamiento; Universidad del Valle: Cali, Colombia, 2011. [Google Scholar]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- ISO 10993-5. Biological Evaluation of Medical Devices Part 5: Tests for Cytotoxicity: In Vitro Methods; International Organization for Standardization: Geneva, Switzerland, 2012. [Google Scholar]
- ISO 10993-12. Biological Evaluation of Medical Devices Part-12: Sample Preparation and Reference Materials; International Organization for Standardization: Geneva, Switzerland, 2012. [Google Scholar]
- Mostafa, N.Y.; Hassan, H.M.; Mohamed, F.H. Sintering behavior and thermal stability of Na+, SiO44− and CO32−co-substituted hydroxyapatites. J. Alloys Compd. 2009, 479, 692–698. [Google Scholar] [CrossRef]
- Zhuang, Z.; Miki, T.; Yumoto, M.; Konishi, T.; Aizawa, M. Ultrastructural observation of hydroxyapatite ceramics with preferred orientation to a-plane using high-resolution transmission electron microscopy. Proced. Eng. 2012, 36, 121–127. [Google Scholar] [CrossRef]
- Voltolini, M.; Wenk, H.R.; Gomez Barreiro, J.; Agarwal, S.C. Hydroxylapatite lattice preferred orientation in bone: A study of macaque, human and bovine samples. J. Appl. Crystallogr. 2011, 44, 928–934. [Google Scholar] [CrossRef]
- Marković, S.; Veselinović, L.; Lukić, M.J.; Karanović, L.; Bračko, I.; Ignjatović, N.; Uskoković, D. Synthetical bone-like and biological hydroxyapatites: a comparative study of crystal structure and morphology. Biomed. Mater. 2011, 6, 45005. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Aizawa, M. Protein adsorption on single-crystal hydroxyapatite particles with preferred orientation to a(b)- And c-axes. J. Mater. Sci. Mater. Med. 2013, 24, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Fujimi, T.J.; Nakamura, M.; Konishi, T.; Yoshimura, H.; Aizawa, M. Development of a,b-plane-oriented hydroxyapatite ceramics as models for living bones and their cell adhesion behavior. Acta Biomater. 2013, 9, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Elliott, J.C.; Dowker, S.E.P.; Rodriguez-Lorenzo, L.M. Rietveld refinements and spectroscopic studies of the structure of Ca-deficient apatite. Biomaterials 2005, 26, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Dowker, S.E.P.; Elliott, J.C. Rietveld refinements and spectroscopic structural studies of a Na-free carbonate apatite made by hydrolysis of monetite. Biomaterials 2006, 27, 4682–4692. [Google Scholar] [CrossRef] [PubMed]
- ISO 13779-1. Implants for Surgery-Hydroxyapatite Part 1: Ceramic Hydroxyapatite; International Organization for Standardization: Geneva, Switzerland, 2008. [Google Scholar]
- Penel, G.; Leroy, G.; Rey, C.; Bres, E. MicroRaman Spectral Study of the PO 4 and CO 3 Vibrational Modes in Synthetic and Biological Apatites. Calcif. Tissue Int. 1998, 63, 475–481. [Google Scholar] [CrossRef]
- Antonakos, A.; Liarokapis, E.; Leventouri, T. Micro-Raman and FTIR studies of synthetic and natural apatites. Biomaterials 2007, 28, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ng, B.; Khor, K.; Cheang, P.; Clyne, T. Raman spectroscopy determination of phases within thermal sprayed hydroxyapatite splats and subsequent in vitro dissolution examination. Acta Mater. 2004, 52, 445–453. [Google Scholar] [CrossRef]
- Carden, A.; Morris, M.D. Application of vibrational spectroscopy to the study of mineralized tissues (review ). J. Biomed. Opt. 2017, 5, 259–268. [Google Scholar] [CrossRef]
- Kim, H.M.; Himeno, T.; Kokubo, T.; Nakamura, T. Process and kinetics of bonelike apatite formation on sintered hydroxyapatite in a simulated body fluid. Biomaterials 2005, 26, 4366–4373. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, M.; Matsuura, T.; Zhuang, Z. Syntheses of single-crystal apatite particles with preferred orientation to the a- and c-axes as models of hard tissue and their applications. Biol. Pharm. Bull. 2013, 36, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, W.; Gibson, I.R. Novel synthesis and characterization of an AB-type carbonate-substituted hydroxyapatite. J. Biomed. Mater. Res. 2002, 59, 697–708. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (Sintered at 1200 °C) | Open Porosity (%) | Vickers Hardness (HV) | Grain Size (µm) |
|---|---|---|---|
| BHAp green sample | 35.04 ± 0.08 | ----- | ----- |
| BHAp 2 h | 16.06 ± 0.25 | 227.78 ± 28.80 | 1.18 ± 0.14 |
| BHAp 4 h | 12.09 ± 1.12 | 332.30 ± 77.40 | 1.53 ± 0.18 |
| CHAp green sample | 35.11 ± 0.23 | ---- | ----- |
| CHAp 2 h | 30.40 ± 2.85 | 80.21 ± 10.46 | 0.72 ± 0.06 |
| CHAp 4 h | 27.17 ± 0.39 | 109.40 ± 19.70 | 0.89 ± 0.06 |
| Source | Sample | a (Å) | c (Å) | Volume (Å3) | Ca/P Rietveld | Ca/P ICP | χ2 |
|---|---|---|---|---|---|---|---|
| Theoretical | 9.432 | 6.881 | 528.8 | 1.67 | - | - | |
| BHAp | Powder | 9.423 | 6.886 | 529.8 | 1.60 | 1.57 | 2.61 |
| 2h | 9.421 | 6.891 | 529.6 | 1.63 | --- | 1.75 | |
| 4h | 9.420 | 6.885 | 529.1 | 1.59 | --- | 2.15 | |
| CHAp | Powder | 9.426 | 6.893 | 530.4 | 1.66 | 1.63 | 2.25 |
| 2h | 9.425 | 6.894 | 530.4 | 1.69 | --- | 2.45 | |
| 4h | 9.425 | 6.892 | 530.2 | 1.65 | --- | 2.36 | |
| Element | BHAp (ppm) | CHAp (ppm) | Values accepted ISO 13779-1:2008 (ppm) |
|---|---|---|---|
| As | 0.00 | 0.09 | <3 |
| Cd | 0.00 | 0.00 | <5 |
| Pb | 0.02 | 0.03 | <30 |
| Hg | 0.00 | 0.00 | <5 |
| Assignment | Reported [33,34,35,36] | Experimental Values (Powders) | |||||
|---|---|---|---|---|---|---|---|
| Stoichiometric HAp | CAp Type A | Cap Type B | Bone | CaO | BHAp | CHAp | |
| v1PO43− | 964 | 947 | 961 | 961 | 960 | 960 | |
| 957 | |||||||
| v2PO43− | 433 | 440 | 432 | 432 | 429 | 427 | |
| 448 | 445 | 452 | 444 | 442 | |||
| v3PO43− | 1029 | 1018 | 1026 | 1032 | 1026 | 1026 | |
| 1034 | 1033 | ||||||
| 1041 | 1038 | ||||||
| 1048 | 1031 | 1047 | 1044 | 1045 | 1045 | ||
| 1057 | 1053 | ||||||
| 1064 | 1064 | ||||||
| 1077 | 1059 | 1070 | 1071 | 1072 | 1074 | ||
| v4PO43−> | 580 | 579 | 579 | 584 | 579 | 577 | |
| 591 | 589 | 590 | 590 | 589 | 589 | ||
| 607 | 608 | 609 | 611 | 605 | 604 | ||
| 614 | 613 | ||||||
| Type A v1CO32− | 1107 | 1103 | 1109 | ||||
| OH stretch | 3573 | 3576 | NO | 3571 | 3570 | ||
| CaO | 1500 | 1500 | |||||
| 1550 | 1545 | ||||||
| 1772 | 1771 | ||||||
| 1935 | 1930 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rincón-López, J.A.; Hermann-Muñoz, J.A.; Giraldo-Betancur, A.L.; De Vizcaya-Ruiz, A.; Alvarado-Orozco, J.M.; Muñoz-Saldaña, J. Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison. Materials 2018, 11, 333. https://doi.org/10.3390/ma11030333
Rincón-López JA, Hermann-Muñoz JA, Giraldo-Betancur AL, De Vizcaya-Ruiz A, Alvarado-Orozco JM, Muñoz-Saldaña J. Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison. Materials. 2018; 11(3):333. https://doi.org/10.3390/ma11030333
Chicago/Turabian StyleRincón-López, July Andrea, Jennifer Andrea Hermann-Muñoz, Astrid Lorena Giraldo-Betancur, Andrea De Vizcaya-Ruiz, Juan Manuel Alvarado-Orozco, and Juan Muñoz-Saldaña. 2018. "Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison" Materials 11, no. 3: 333. https://doi.org/10.3390/ma11030333
APA StyleRincón-López, J. A., Hermann-Muñoz, J. A., Giraldo-Betancur, A. L., De Vizcaya-Ruiz, A., Alvarado-Orozco, J. M., & Muñoz-Saldaña, J. (2018). Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison. Materials, 11(3), 333. https://doi.org/10.3390/ma11030333