Study of Electronic Structure, Thermal Conductivity, Elastic and Optical Properties of α, β, γ-Graphyne


Abstract
1. Introduction
2. Calculation and Models
3. Results and Discussion
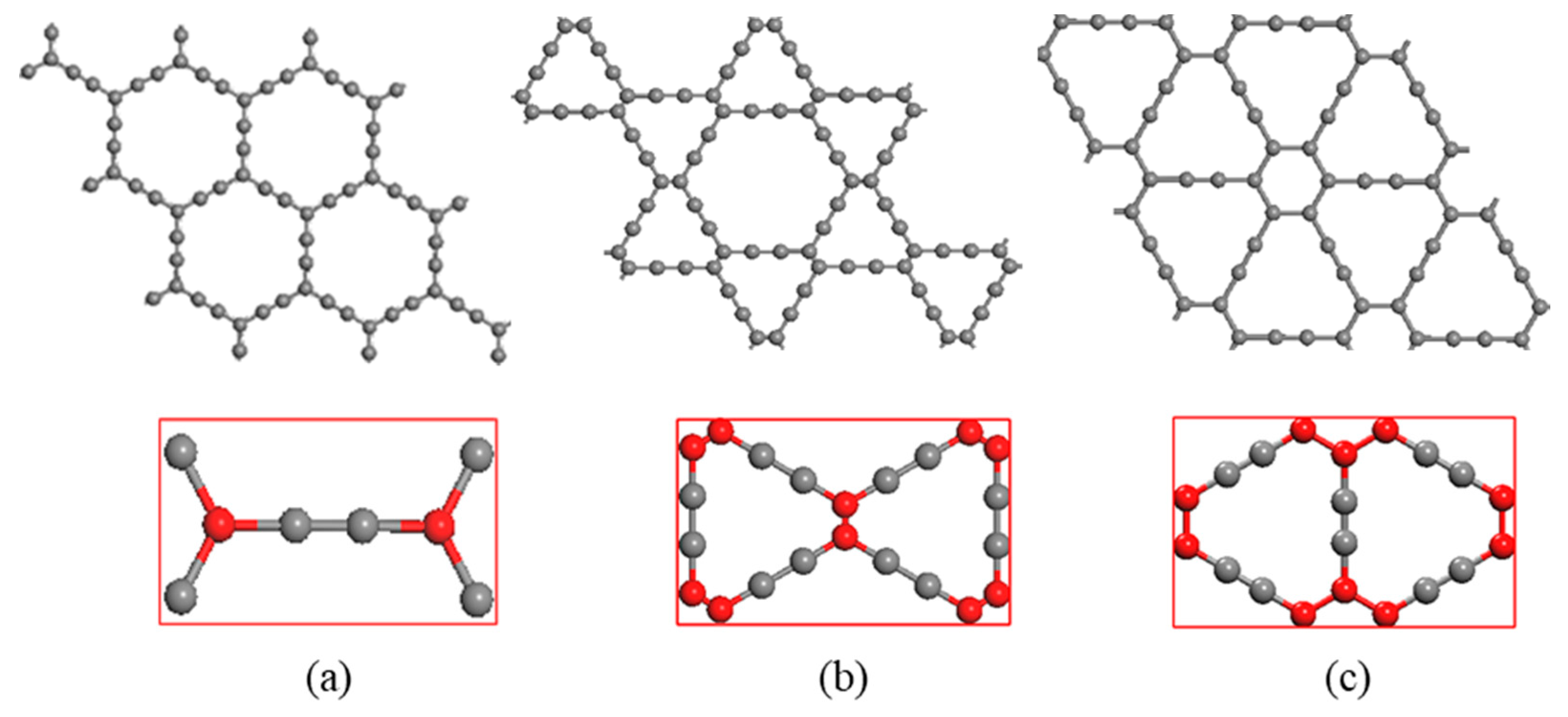
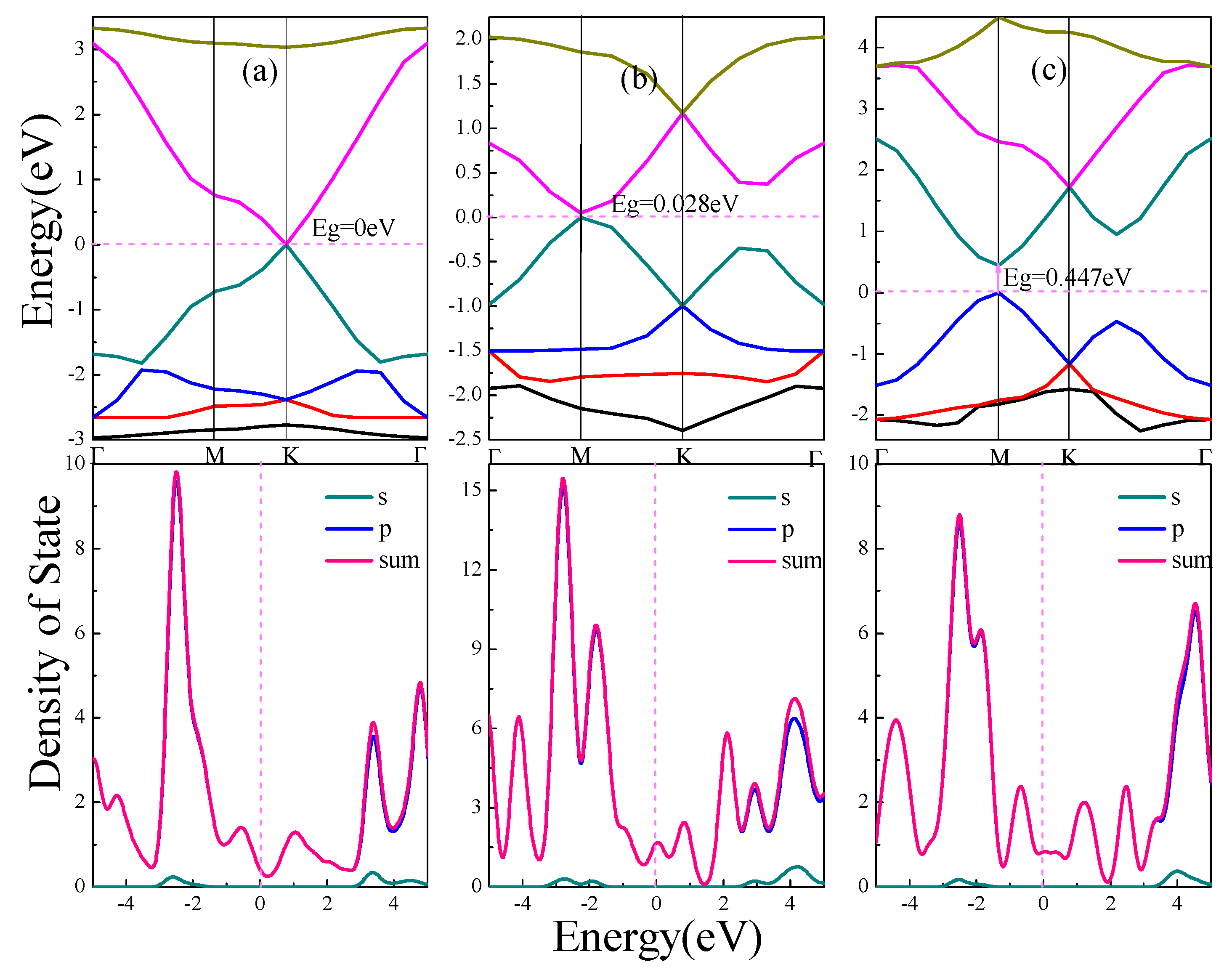
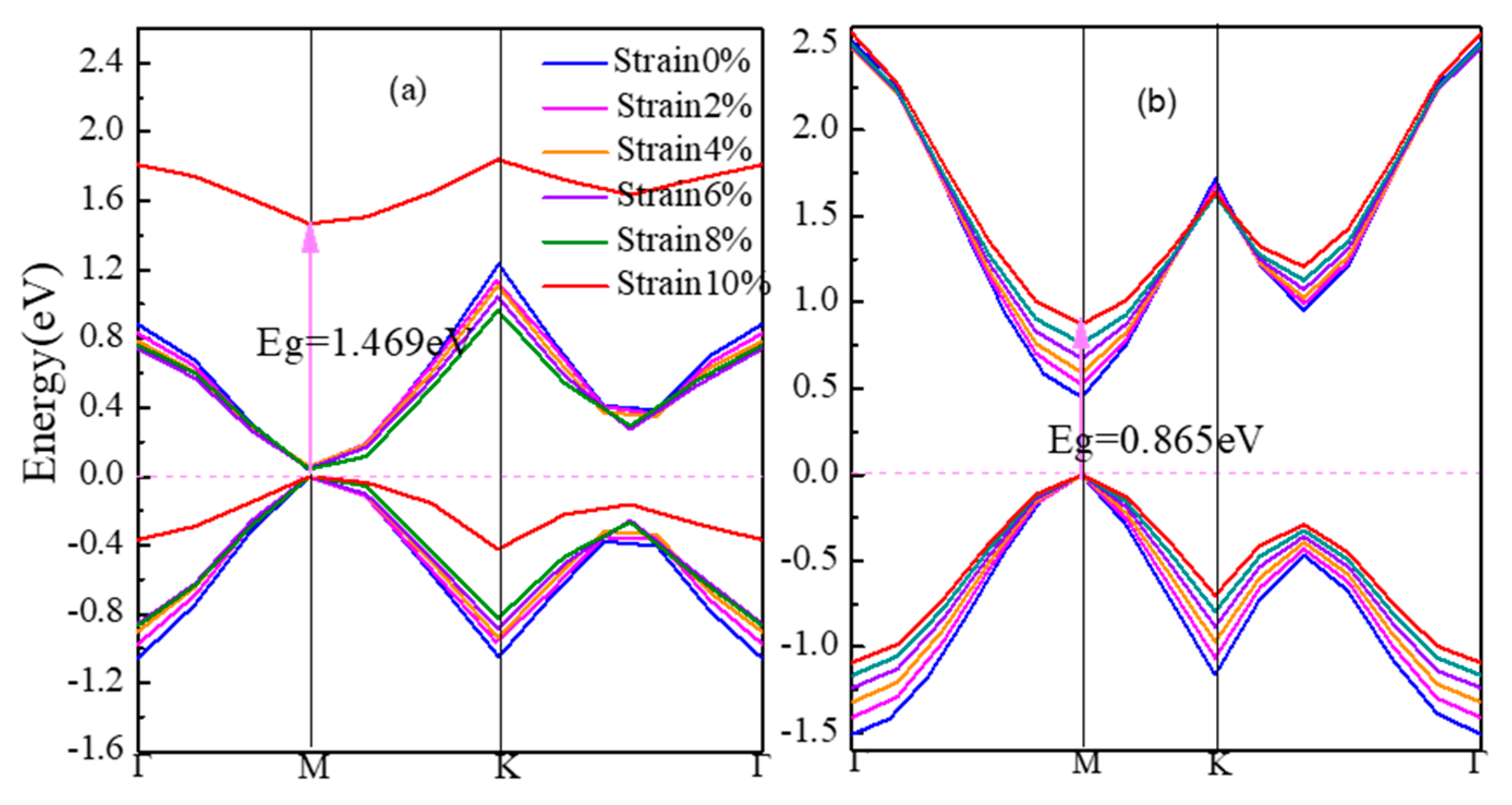
3.1. Electronic Structures
3.2. Elasticity
3.3. Thermal Conductivity
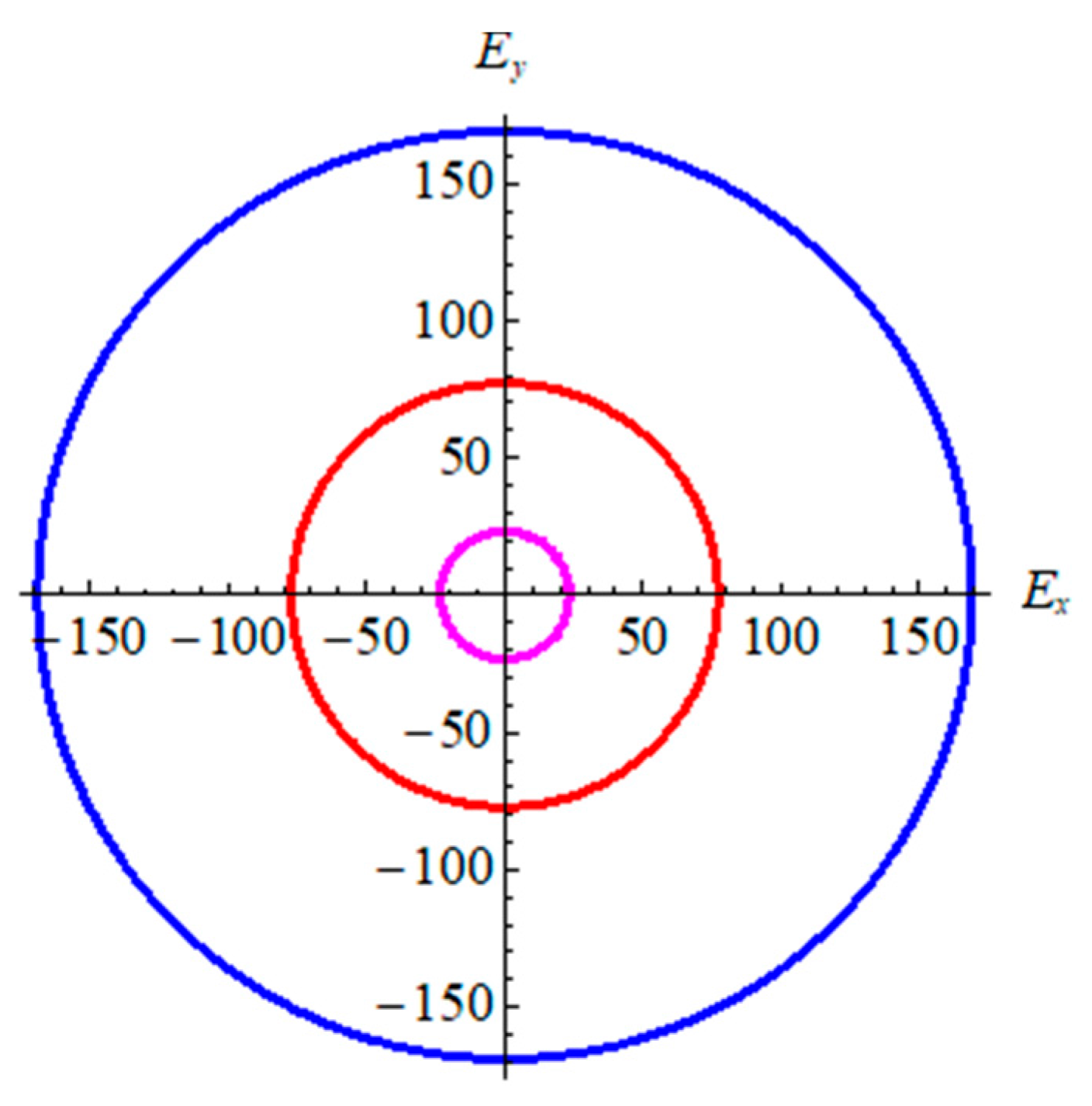
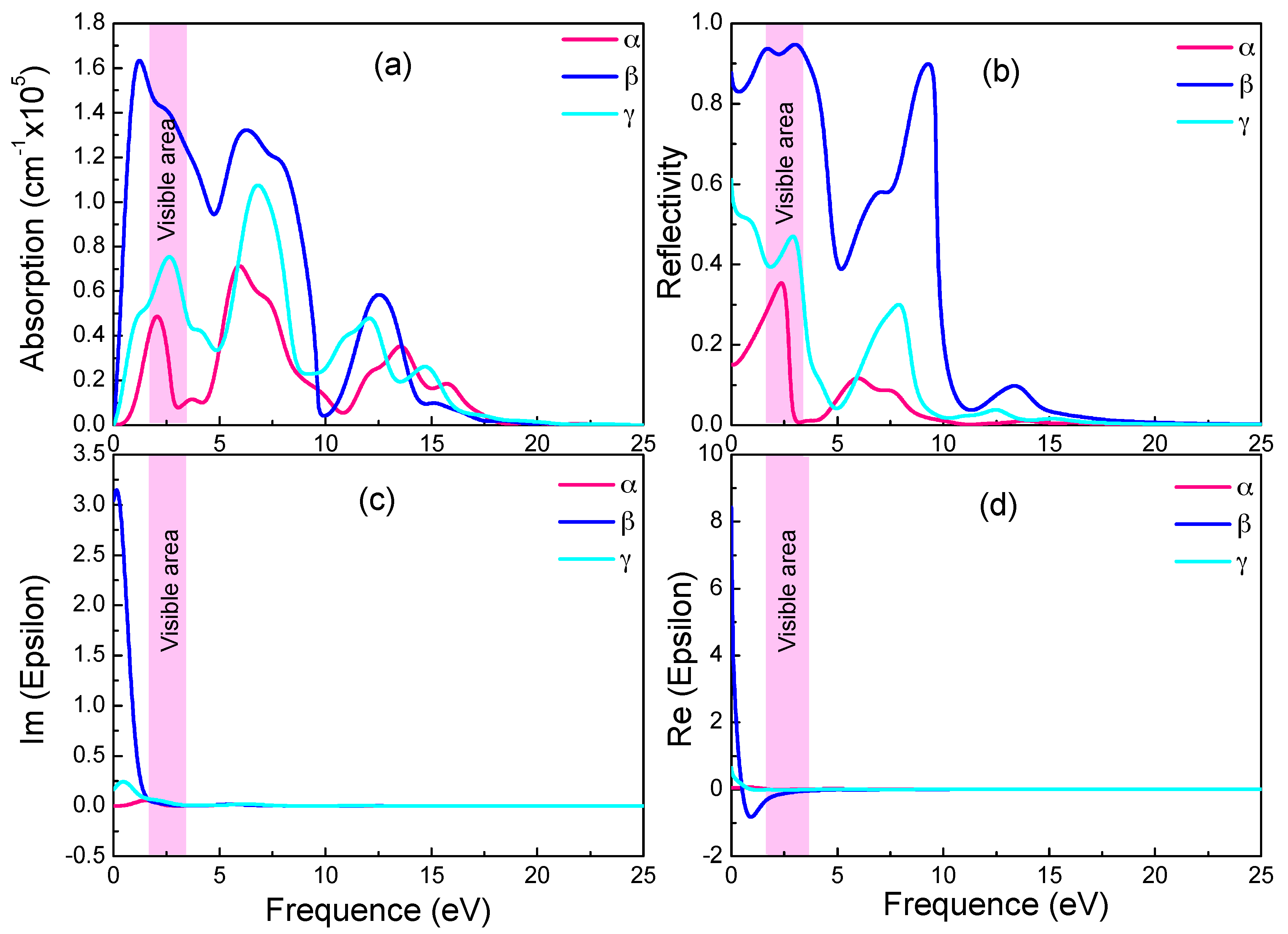
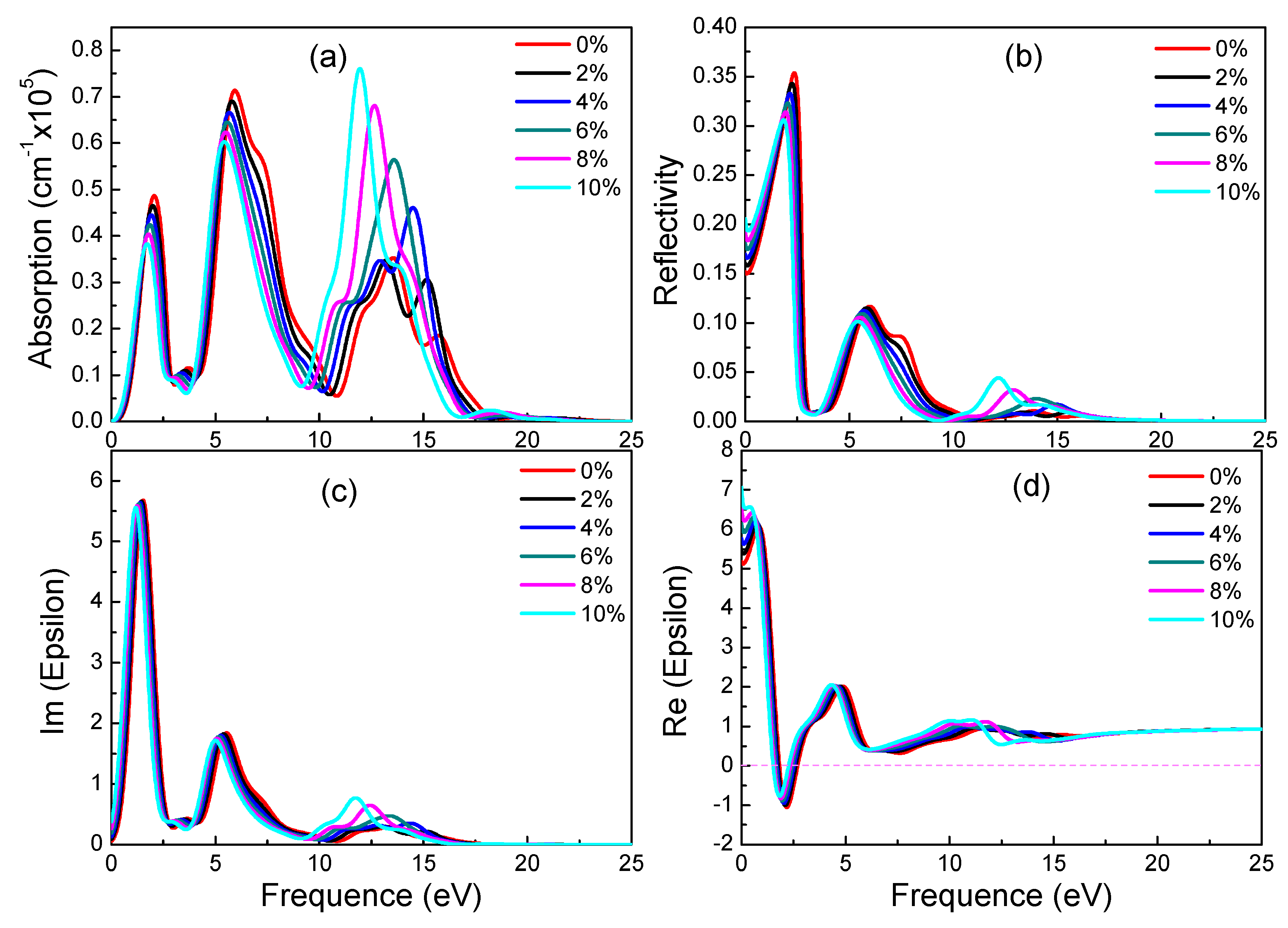
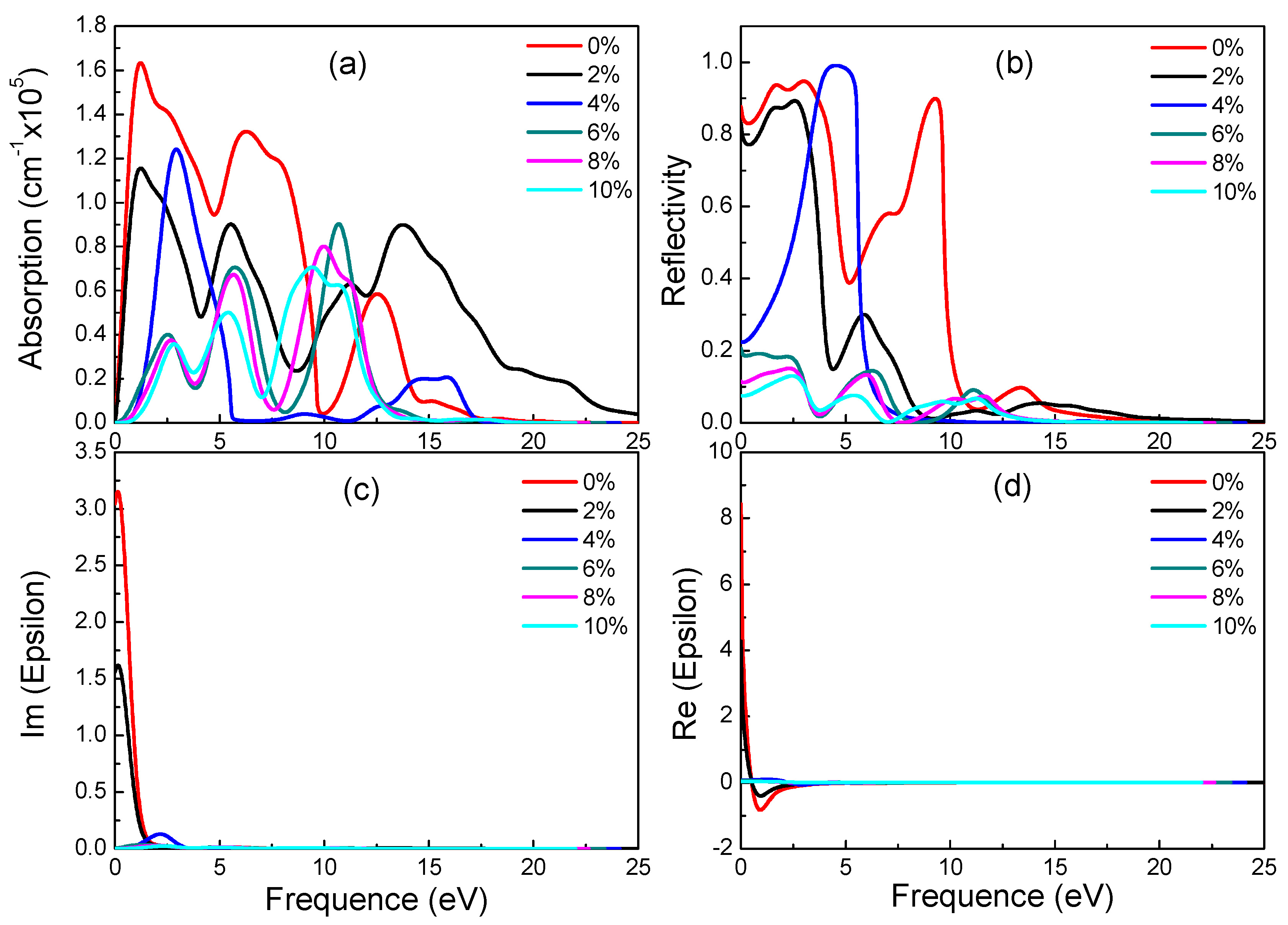
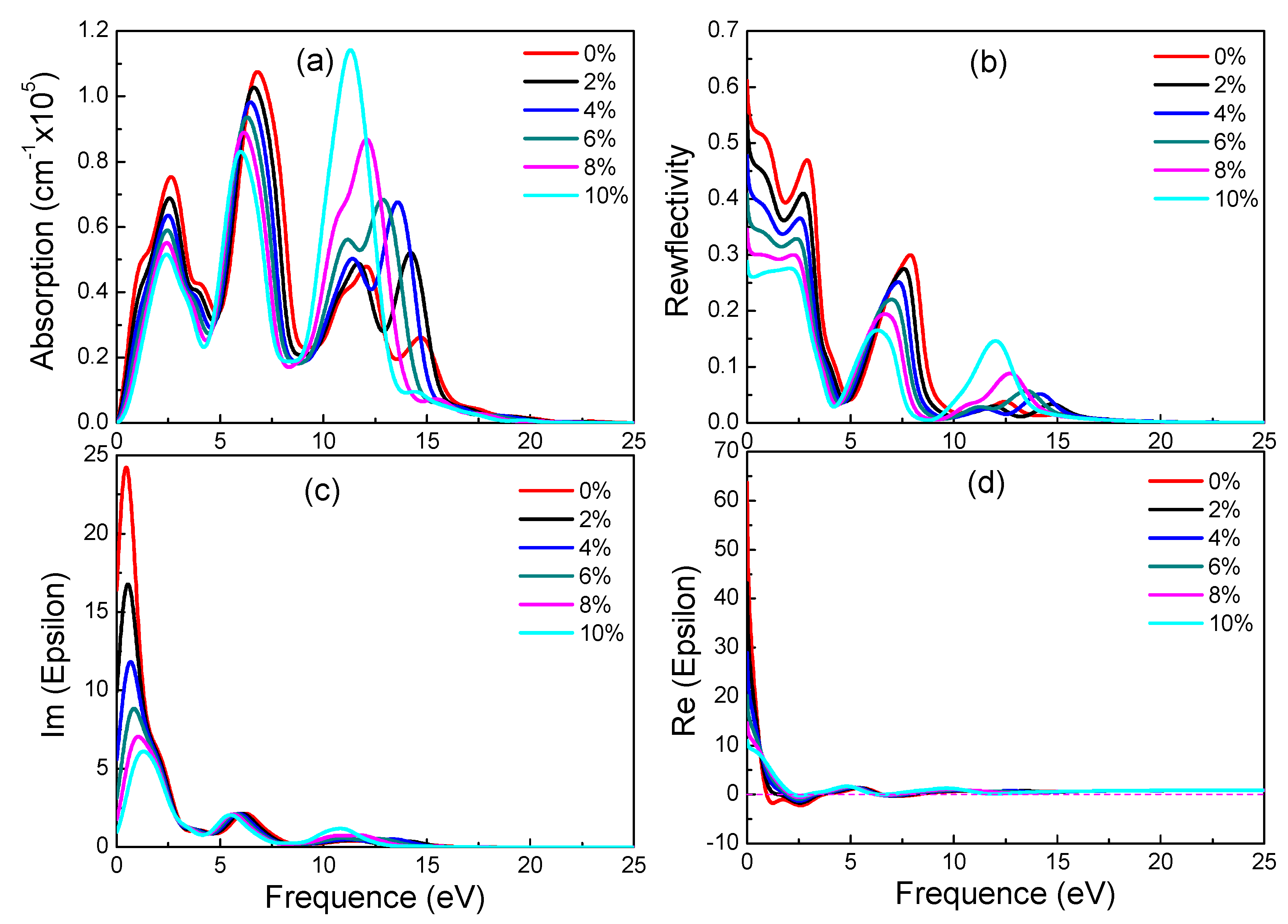
3.4. Optical Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peierls, R. Quelques propriétés typiques des corps solides. Annales de L’institut Henri Poincaré 1935, 5, 177–222. [Google Scholar]
- Landau, L.; Teller, E. Zur Theorie Der Schalldispersion. Physikalische Zeitschrift der Sowjetunion 1936, 10, 34. [Google Scholar]
- Mermin, N.D. Erratum: Crystalline order in two dimensions. Phys. Rev. B 1968, 20, 4762. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Small, J.P.; Amori, M.E.S.; Kim, P. Electric field modulation of galvanomagnetic properties of mesoscopic graphite. Phys. Rev. Lett. 2005, 94, 176803. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Song, Z.M.; Li, T.B.; Li, X.B.; Ogbazghi, A.Y.; Feng, R.; Dai, Z.T.; Marchenkov, A.N.; Conrad, E.H.; First, P.N.; et al. Ultrathin epitaxial graphite: 2D electron gas properties and a route toward graphene-based nanoelectronics. J. Phys. Chem. B 2004, 108, 19912–19916. [Google Scholar] [CrossRef]
- Bunch, J.S.; Yaish, Y.; Brink, M.; Bolotin, K.; McEuen, P.L. Coulomb oscillations and Hall effect in quasi-2D graphite quantum dots. Nano Lett. 2005, 5, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Emtsev, K.V.; Bostwick, A.; Horn, K.; Jobst, J.; Kellogg, G.L.; Ley, L.; McChesney, J.L.; Ohta, T.; Reshanov, S.A.; Rohrl, J.; et al. Towards wafer-size graphene layers by atmospheric pressure graphitization of silicon carbide. Nat. Mater. 2009, 8, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K. Graphene: Status and Prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Malko, D.; Neiss, C.; Vines, F.; Gorling, A. Competition for Graphene: Graphynes with Direction-Dependent Dirac Cones. Phys. Rev. Lett. 2012, 108, 086804. [Google Scholar] [CrossRef] [PubMed]
- Li, G.X.; Li, Y.L.; Liu, H.B.; Guo, Y.B.; Li, Y.J.; Zhu, D.B. Architecture of graphdiyne nanoscale films. Chem. Commun. 2010, 46, 3256–3258. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, H.C.; Zhang, X.M.; Liu, X.B.; Zhao, M.W. Dirac cones and highly anisotropic electronic structure of super-graphyne. Carbon 2017, 113, 40–45. [Google Scholar] [CrossRef]
- Hu, M.; Ma, M.; Zhao, Z. Superhard sp2–sp3 hybrid carbon allotropes with tunable electronic properties. AIP Adv. 2016, 6, 055020. [Google Scholar] [CrossRef]
- He, C.; Sun, L.; Zhang, C.; Zhong, J. Two viable three-dimensional carbon semiconductors with an entirely sp2 configuration. Phys. Chem. Chem. Phys. 2013, 15, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, X.; Zhao, M.; Zhang, M.; Zhao, L.; Feng, X.; Luo, Y. Tunable Hydrogen Separation in sp–sp2 Hybridized Carbon Membranes: A First-Principles Prediction. J. Phys. Chem. C 2012, 116, 16634–16638. [Google Scholar] [CrossRef]
- Kang, J.; Li, J.B.; Wu, F.M.; Li, S.S.; Xia, J.B. Elastic, Electronic, and Optical Properties of Two-Dimensional Graphyne Sheet. J. Phys. Chem. C 2011, 115, 20466–20470. [Google Scholar] [CrossRef]
- Owens, F.J. On the possibility of planar graphyne and graphdiyne chains. Solid State Commun. 2017, 250, 75–78. [Google Scholar] [CrossRef]
- Puigdollers, A.R.; Alonso, G.; Gamallo, P. First-principles study of structural, elastic and electronic properties of alpha-, beta- and gamma-graphyne. Carbon 2016, 96, 879–887. [Google Scholar] [CrossRef]
- Jiang, P.H.; Liu, H.J.; Cheng, L.; Fan, D.D.; Zhang, J.; Wei, J.; Liang, J.H.; Shi, J. Thermoelectric properties of gamma-graphyne from first-principles calculations. Carbon 2017, 113, 108–113. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Singh, N.B.; Sarkar, U. Pristine and BN doped graphyne derivatives for UV light protection. Int. J. Quantum Chem. 2015, 115, 820–829. [Google Scholar] [CrossRef]
- Sarma, J.V.N.; Chowdhury, R.; Jayaganthan, R. Graphyne-Based Single Electron Transistor: Ab Initio Analysis. Nano 2014, 9, 1450032. [Google Scholar] [CrossRef]
- Wu, P.; Du, P.; Zhang, H.; Cai, C.X. Graphyne-supported single Fe atom catalysts for CO oxidation. Phys. Chem. Chem. Phys. 2015, 17, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.F.; Rao, D.W.; Meng, Z.S.; Zhang, X.B.; Xu, G.J.; Liu, Y.Z.; Kan, E.J.; Xiao, C.Y.; Deng, K.M. Boron-substituted graphyne as a versatile material with high storage capacities of Li and H-2: A multiscale theoretical study. Phys. Chem. Chem. Phys. 2013, 15, 16120–16126. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, W.B.; Wang, R.G.; Hao, L.F.; Jiao, W.C. Hydrogen storage using Na-decorated graphyne and its boron nitride analog. Int. J. Hydrogen Energy 2014, 39, 12757–12764. [Google Scholar] [CrossRef]
- Ozcelik, V.O.; Ciraci, S. Size Dependence in the Stabilities and Electronic Properties of alpha-Graphyne and Its Boron Nitride Analogue. J. Phys. Chem. C 2013, 117, 2175–2182. [Google Scholar] [CrossRef]
- Wang, G.X.; Si, M.S.; Kumar, A.; Pandey, R. Strain engineering of Dirac cones in graphyne. Appl. Phys. Lett. 2014, 104, 213107. [Google Scholar] [CrossRef]
- Long, M.Q.; Tang, L.; Wang, D.; Li, Y.L.; Shuai, Z.G. Electronic Structure and Carrier Mobility in Graphdiyne Sheet and Nanoribbons: Theoretical Predictions. ACS Nano 2011, 5, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.D.; Zhang, L.Z.; Song, B.Q.; Du, S.X.; Gao, H.J. Graphyne- and graphdiyne-based nanoribbons: Density functional theory calculations of electronic structures. Appl. Phys. Lett. 2011, 98, 173102. [Google Scholar] [CrossRef]
- Srinivasu, K.; Ghosh, S.K. Graphyne and Graphdiyne: Promising Materials for Nanoelectronics and Energy Storage Applications. J. Phys. Chem. C 2012, 116, 5951–5956. [Google Scholar] [CrossRef]
- Ansari, R.; Mirnezhad, M.; Rouhi, H.; Faghihnasiri, M. Young’s modulus and poisson’s ratio of monolayer graphyne. J. Nanostruct. 2013, 3, 303–307. [Google Scholar]
- Shao, Z.-G.; Sun, Z.-L. Optical properties of α-, β-, γ-, and 6,6,12-graphyne structures: First-principle calculations. Phys. E 2015, 74, 438–442. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Pei, Q.X.; Wang, C.M. A molecular dynamics investigation on thermal conductivity of graphynes. Comput. Mater. Sci. 2012, 65, 406–410. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.M.; Alder, B.J. Ground-State of the Electron-Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Broyden, C.G. The Convergence of a Class of Double-rank Minimization Algorithms 1. General Considerations. J. Appl. Math. 1970, 6, 76–90. [Google Scholar] [CrossRef]
- Dickman, S.; Senozan, N.M.; Hunt, R.L. Thermodynamic Properties and the Cohesive Energy of Calcium Ammoniate. J. Chem. Phys. 1970, 52, 2657–2663. [Google Scholar] [CrossRef]
- Chiou, W.C.; Carter, E.A. Structure and stability of Fe3C-cementite surfaces from first principles. Surf. Sci. 2003, 530, 87–100. [Google Scholar] [CrossRef]
- Yue, Q.; Chang, S.; Kang, J.; Tan, J.; Qin, S.; Li, J. Magnetic and electronic properties of α-graphyne nanoribbons. J. Chem. Phys. 2012, 136, 244702. [Google Scholar] [CrossRef] [PubMed]
- Sevincli, H.; Sevik, C. Electronic, phononic, and thermoelectric properties of graphyne sheets. Appl. Phys. Lett. 2014, 105, 223108. [Google Scholar] [CrossRef]
- Peng, Q.; Ji, W.; De, S. Mechanical properties of graphyne monolayers: A first-principles study. Phys. Chem. Chem. Phys. 2012, 14, 13385–13391. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lv, K.; Wang, Q.; Chen, X.S.; Sun, Q.; Jena, P. Electronic structures and bonding of graphyne sheet and its BN analog. J. Chem. Phys. 2011, 134, 174701. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.N.; Hu, M.; Peng, Y.S.; Luo, Y.T.; Li, C.M.; Chen, Z.G. Electronic, elastic, and optical properties of monolayer BC2N. J. Solid State Chem. 2016, 244, 120–128. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-general least-squares extraction of elastic data for strained materials from ab initio calculations of stress. Phys. Rev. B 2002, 65, 104104. [Google Scholar] [CrossRef]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Wilhoit, R.C.; Chao, J.; Hall, K.R. Thermodynamic Properties of Key Organic Oxygen Compounds in the Carbon Range C1 to C4. Part 1. Properties of Condensed Phases. J. Phys. Chem. Ref. Data 1985, 14, 549–550. [Google Scholar] [CrossRef]
- Nika, D.L.; Pokatilov, E.P.; Askerov, A.S.; Balandin, A.A. Phonon thermal conduction in graphene: Role of Umklapp and edge roughness scattering. Phys. Rev. B 2009, 79, 155413. [Google Scholar] [CrossRef]
- Yang, Z.; Ji, Y.L.; Lan, G.Q.; Xu, L.C.; Wang, H.; Liu, X.G.; Xu, B.S. The thermal properties and thermoelectric performance of gamma-graphyne nanoribbons. J. Phys. D Appl. Phys. 2016, 49, 145102. [Google Scholar] [CrossRef]
- Chen, Z.Q.; Li, F.; Hu, M.; Li, C.M. Elastic properties, hardness, and anisotropy in baddeleyite IVTMO2 (M = Ti, Zr, Hf). Sci. China Mater. 2015, 58, 893–905. [Google Scholar] [CrossRef]
- Clarke, D.R. Materials selection guidelines for low thermal conductivity thermal barrier coatings. Surf. Coat. Technol. 2003, 163, 67–74. [Google Scholar] [CrossRef]
- Bouhemadou, A. First-principles study of structural, electronic and elastic properties of Nb4AlC3. Braz. J. Phys. 2010, 40, 52–57. [Google Scholar] [CrossRef]
- Waters, K.R.; Hughes, M.S.; Brandenburger, G.H.; Miller, J.G. On a time-domain representation of the Kramers-Kronig dispersion relations. J. Acoust. Soc. Am. 2000, 108, 2114–2119. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Method | a | ρ2D | sp-sp | sp-sp2 | sp2-sp2 | Band Gap | Etotal | Ecoh |
|---|---|---|---|---|---|---|---|---|---|
| α-graphyne | GGA-PBE | 6.950 | 2.357 | 1.229 | 1.392 | - | 0 | −1233.384 | −8.343 |
| GAA-PBE-D | 6.948 | 2.359 | 1.228 | 1.391 | - | 0.005 | −1233.57 | −8.366 | |
| Other work | 6.966 a, 7.01 b | 1.230 a | 1.396 a | - | - | - | - | ||
| β-graphyne | GGA-PBE | 9.459 | 2.863 | 1.232 | 1.386 | 1.456 | 0.028 | −2776.578 | −8.424 |
| GAA-PBE-D | 9.454 | 2.867 | 1.231 | 1.385 | 1.455 | 0.04 | −2777.185 | −8.458 | |
| Other work | 9.47 c, 9.464 d | 1.232 a | 1.389 a | 1.457 a | - | - | - | ||
| γ-graphyne | GGA-PBE | 6.875 | 3.614 | 1.222 | 1.403 | 1.422 | 0.447 | −1853.464 | −8.625 |
| GAA-PBE-D | 6.870 | 3.619 | 1.221 | 1.403 | 1.422 | 0.448 | −1853.99 | −8.699 | |
| Other work | 6.89 a,6.86 e | 2.357 | 1.223 a | 1.408 a | 1.426 a | 0.46 f | - | −7.95 e |
| Parameters | α-Graphyne | β-Graphyne | γ-Graphyne |
|---|---|---|---|
| C11 | 95 | 133 | 202 |
| C12 | 82 | 86 | 82 |
| C22 | 95 | 133 | 202 |
| C66 | 6.5 | 23.5 | 60 |
| S11 | 0.043 | 0.013 | 0.006 |
| S12 | −0.037 | −0.008 | −0.002 |
| S22 | 0.043 | 0.013 | 0.006 |
| S66 | 0.161 | 0.043 | 0.017 |
| G2D | 6.5 | 23.5 | 60 |
| B2D | 89 | 110 | 142 |
| E2D[10] = E2D[01] | 24 (22 a) | 77 (73 a) | 169 (166 a, 169 b) |
| v2D[10] = v2D[01] | 0.863 (0.87 a) | 0.647 (0.67 a) | 0.406 (0.42 a, 0.417 b) |
| A | 1 | 1 | 1 |
| Species | kmin | θD | vt | vl | vm |
|---|---|---|---|---|---|
| α-Graphyne | 0.920 | 220 | 1.322 | 3.246 | 1.497 |
| β-Graphyne | 1.650 | 307 | 1.853 | 3.823 | 2.083 |
| γ-Graphyne | 2.456 | 410 | 2.274 | 4.082 | 2.532 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Xie, Z.; Li, C.; Li, G.; Chen, Z. Study of Electronic Structure, Thermal Conductivity, Elastic and Optical Properties of α, β, γ-Graphyne. Materials 2018, 11, 188. https://doi.org/10.3390/ma11020188
Hou X, Xie Z, Li C, Li G, Chen Z. Study of Electronic Structure, Thermal Conductivity, Elastic and Optical Properties of α, β, γ-Graphyne. Materials. 2018; 11(2):188. https://doi.org/10.3390/ma11020188
Chicago/Turabian StyleHou, Xun, Zhongjing Xie, Chunmei Li, Guannan Li, and Zhiqian Chen. 2018. "Study of Electronic Structure, Thermal Conductivity, Elastic and Optical Properties of α, β, γ-Graphyne" Materials 11, no. 2: 188. https://doi.org/10.3390/ma11020188
APA StyleHou, X., Xie, Z., Li, C., Li, G., & Chen, Z. (2018). Study of Electronic Structure, Thermal Conductivity, Elastic and Optical Properties of α, β, γ-Graphyne. Materials, 11(2), 188. https://doi.org/10.3390/ma11020188