Fingerprints of sp1 Hybridized C in the Near-Edge X-ray Absorption Spectra of Surface-Grown Materials

 , , , , , ,
, , , , , , 

Abstract
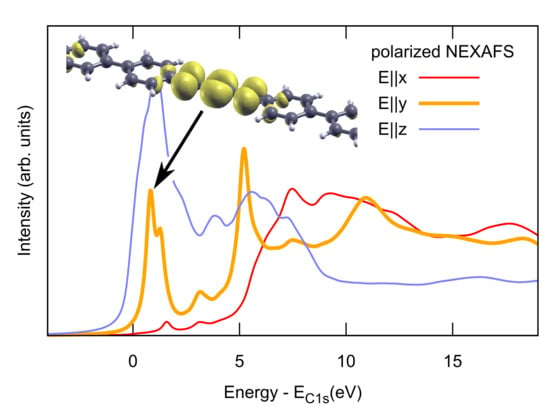
{kind=link}
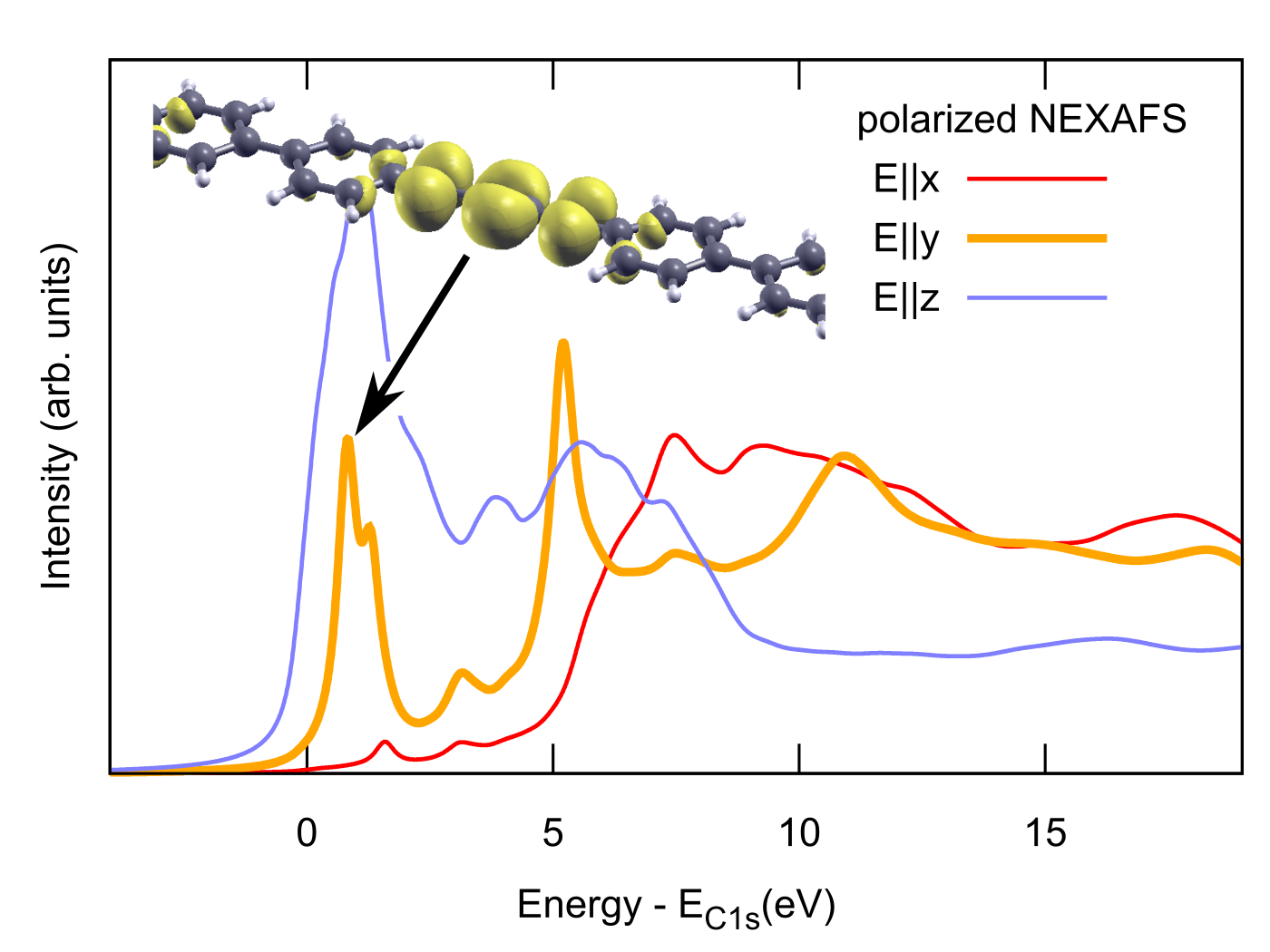
{kind=link}
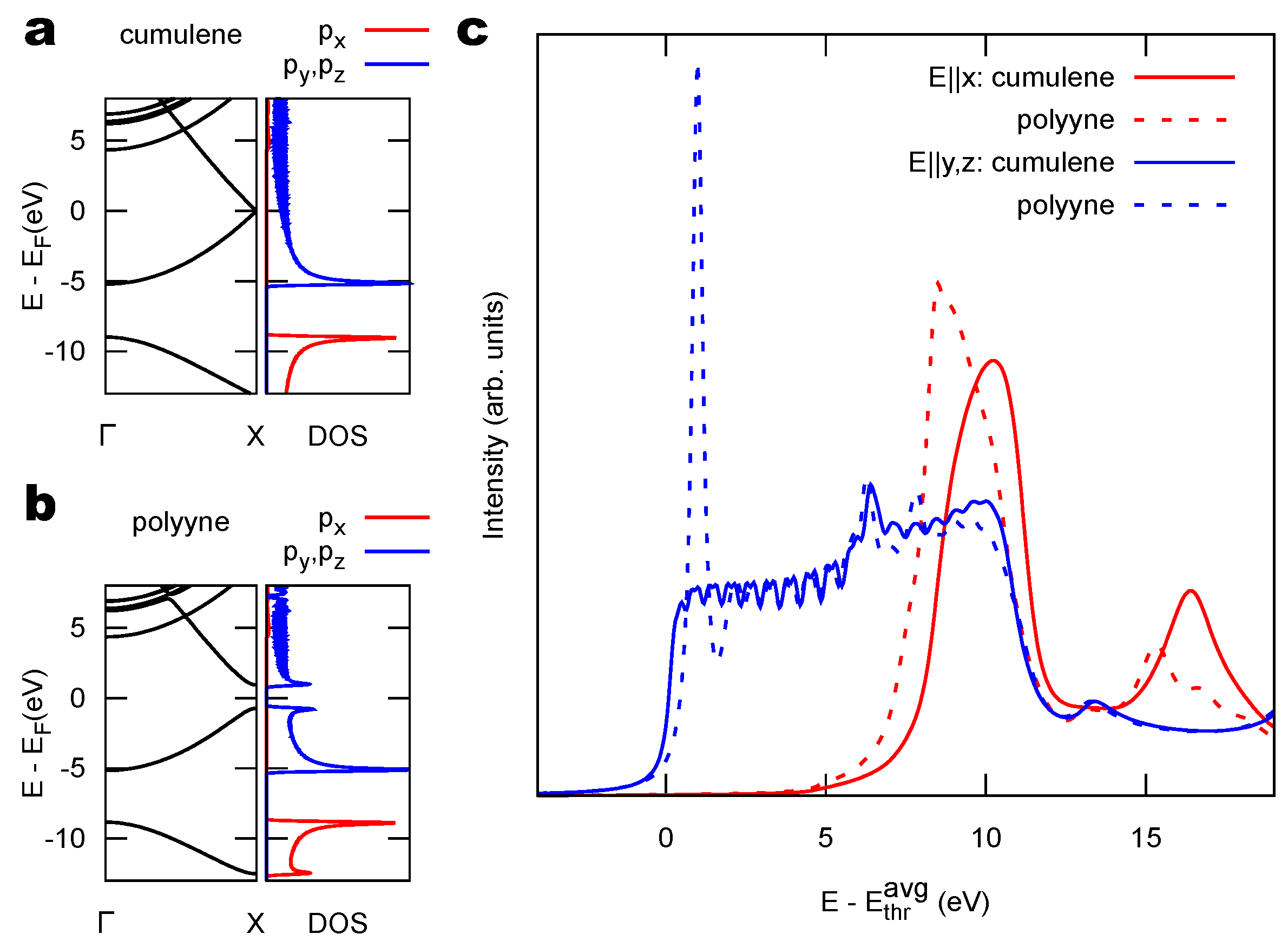
{kind=link}
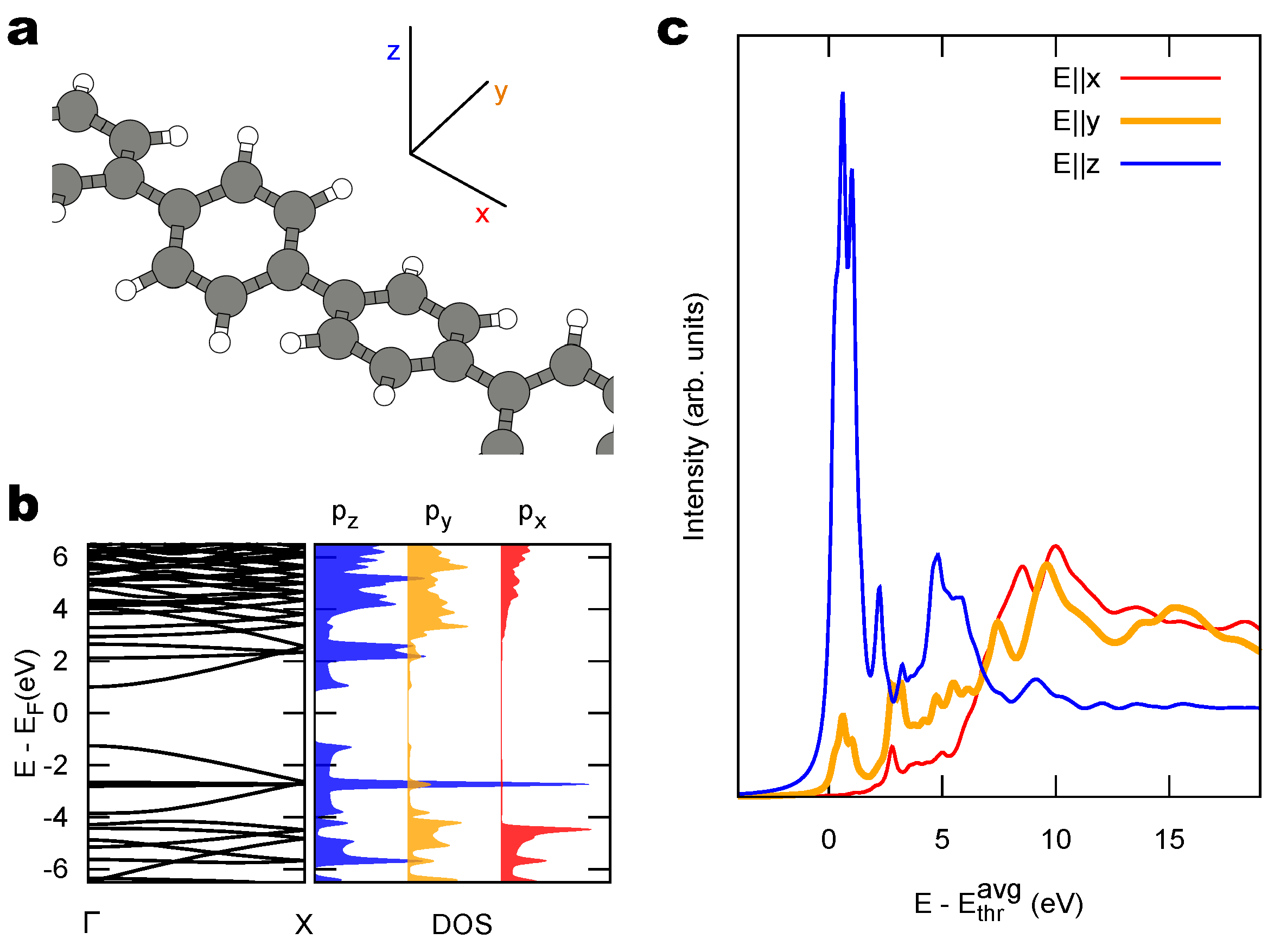
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
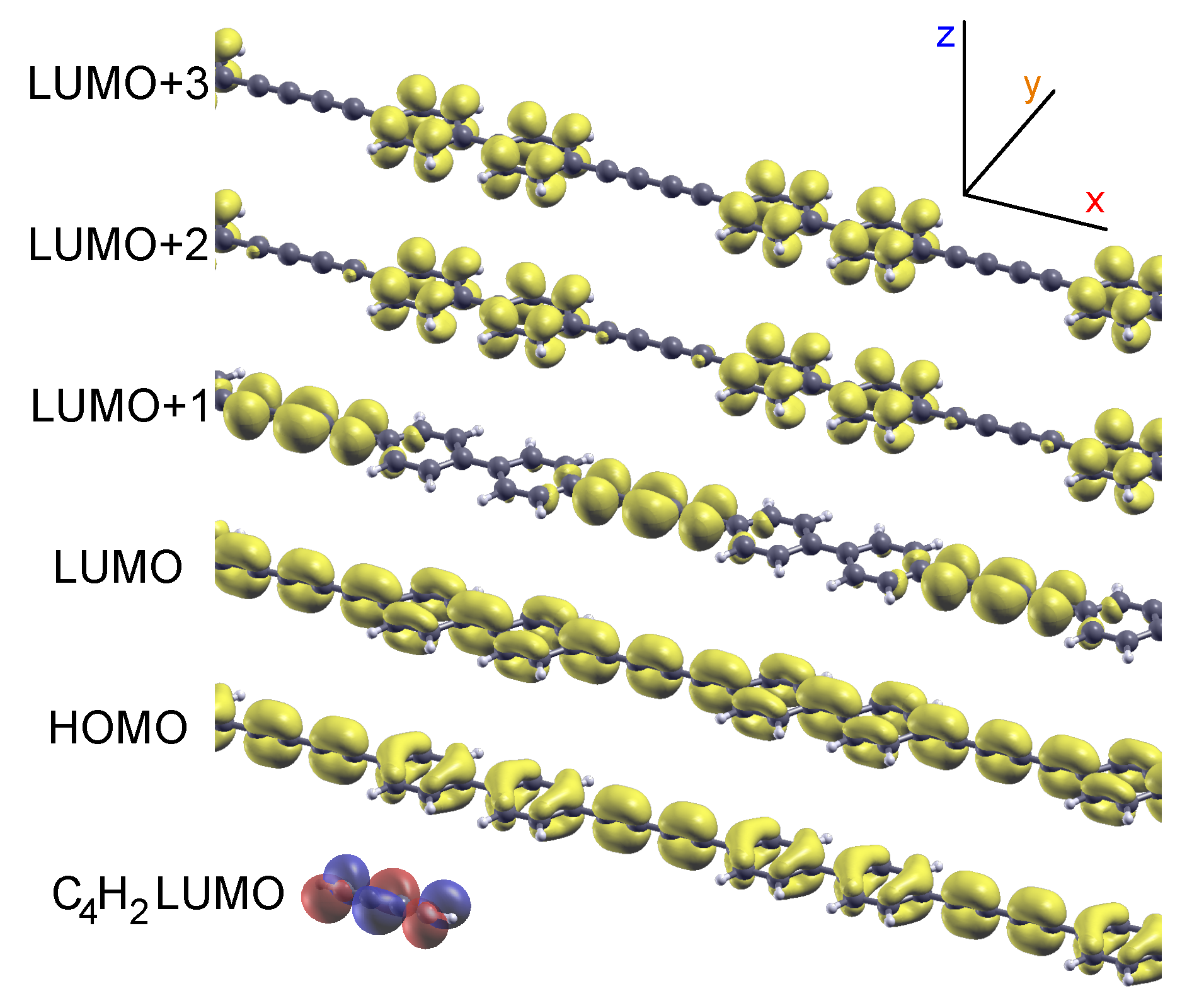
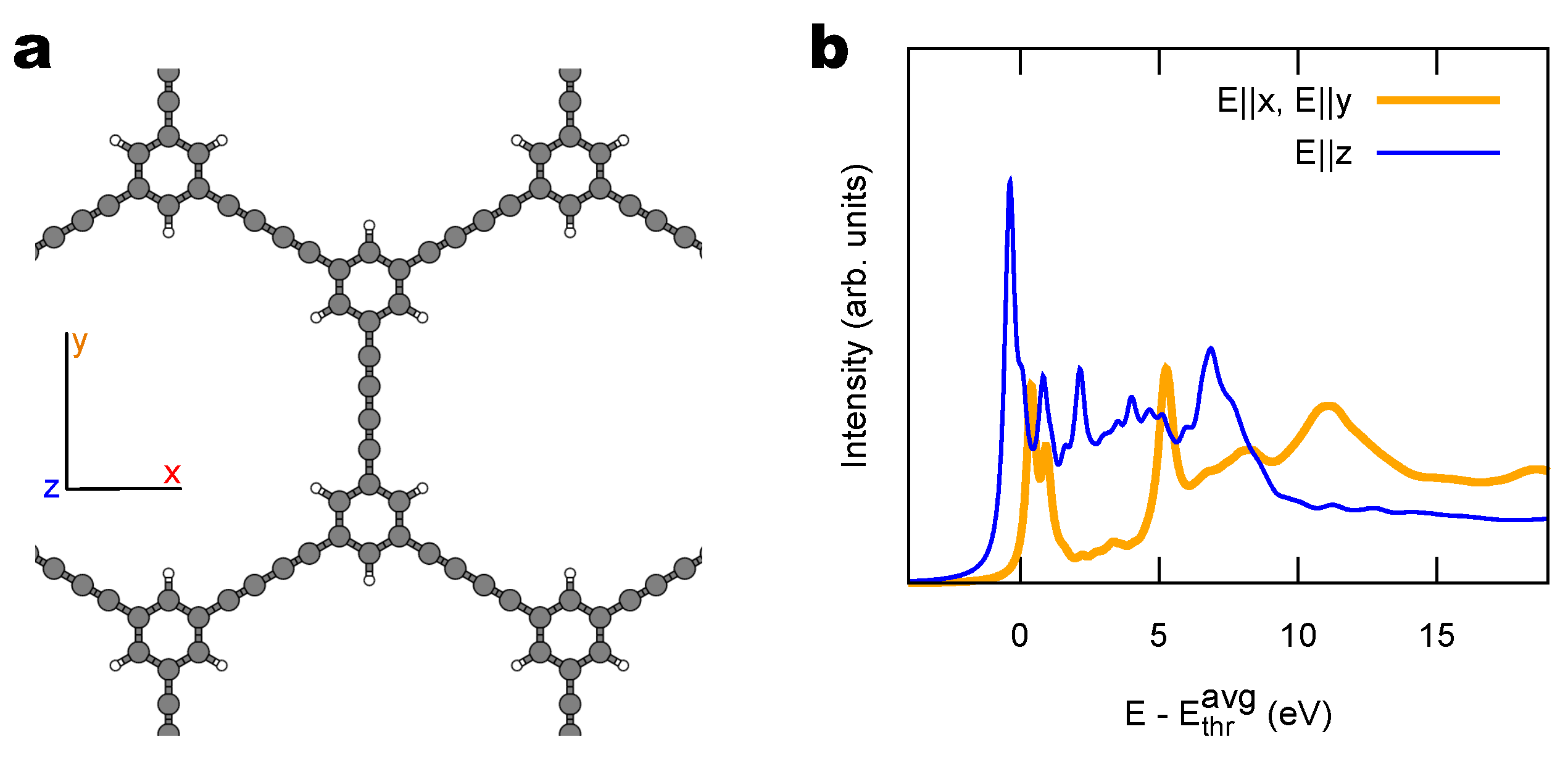
2.1. Model 1D Systems
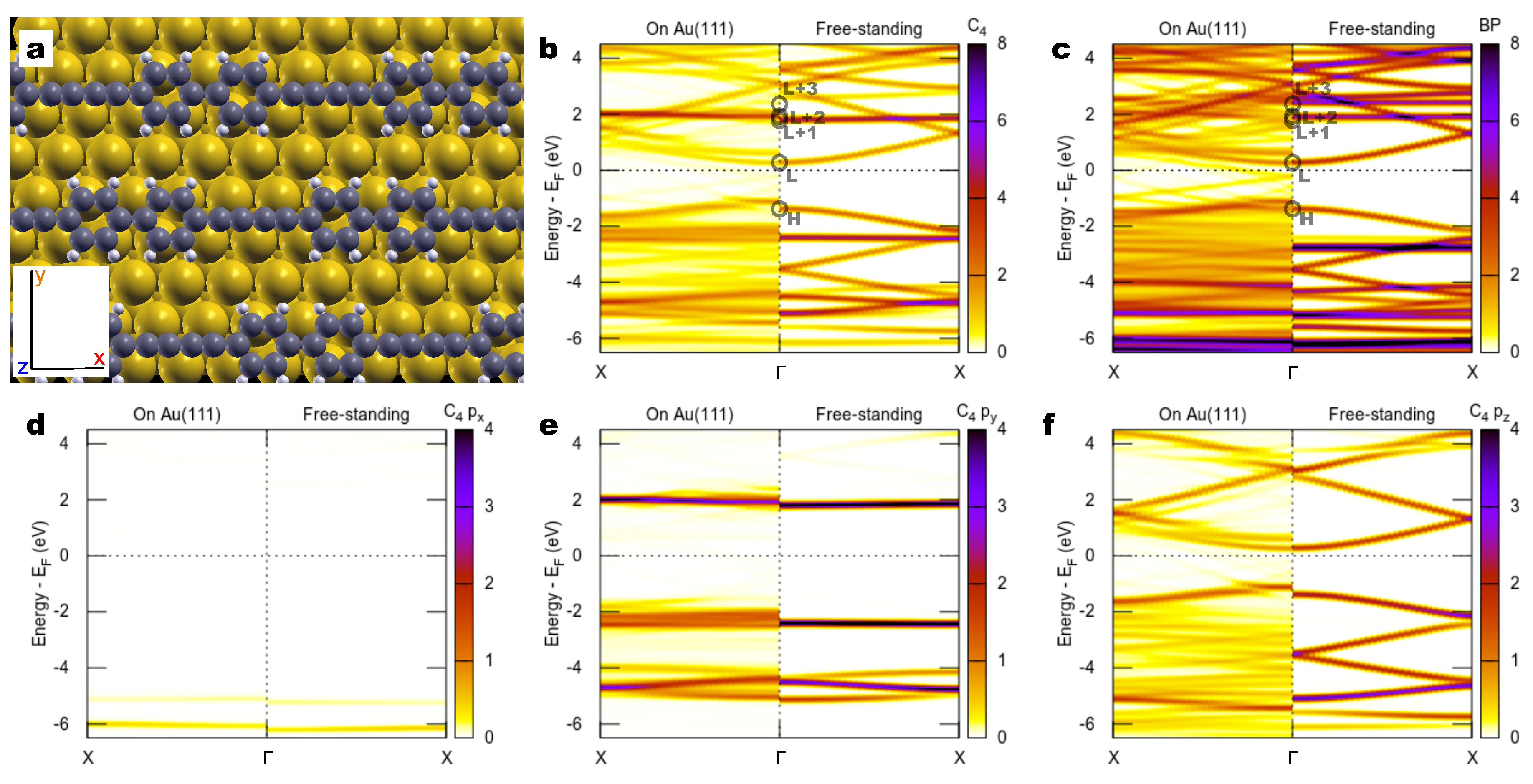
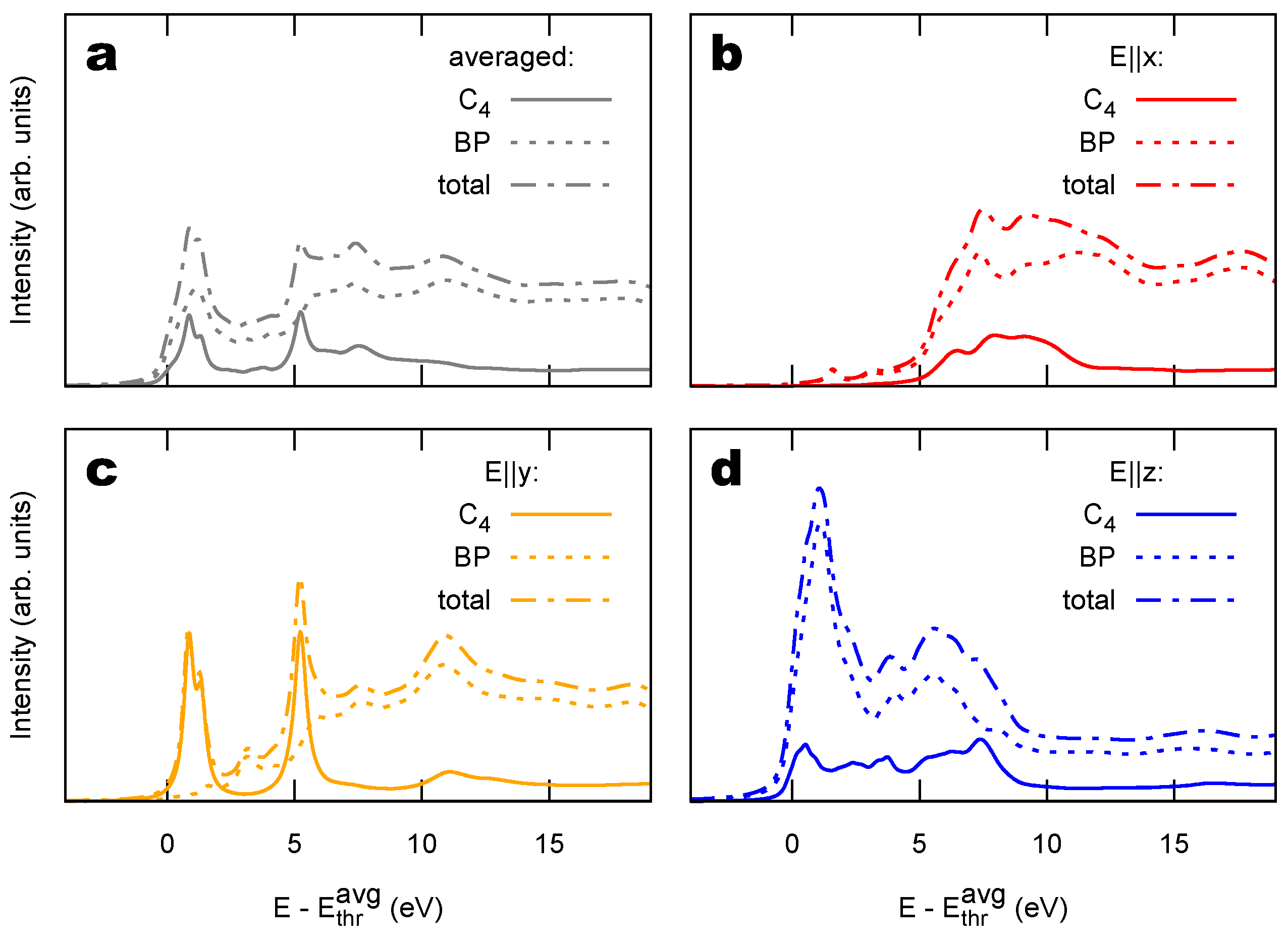
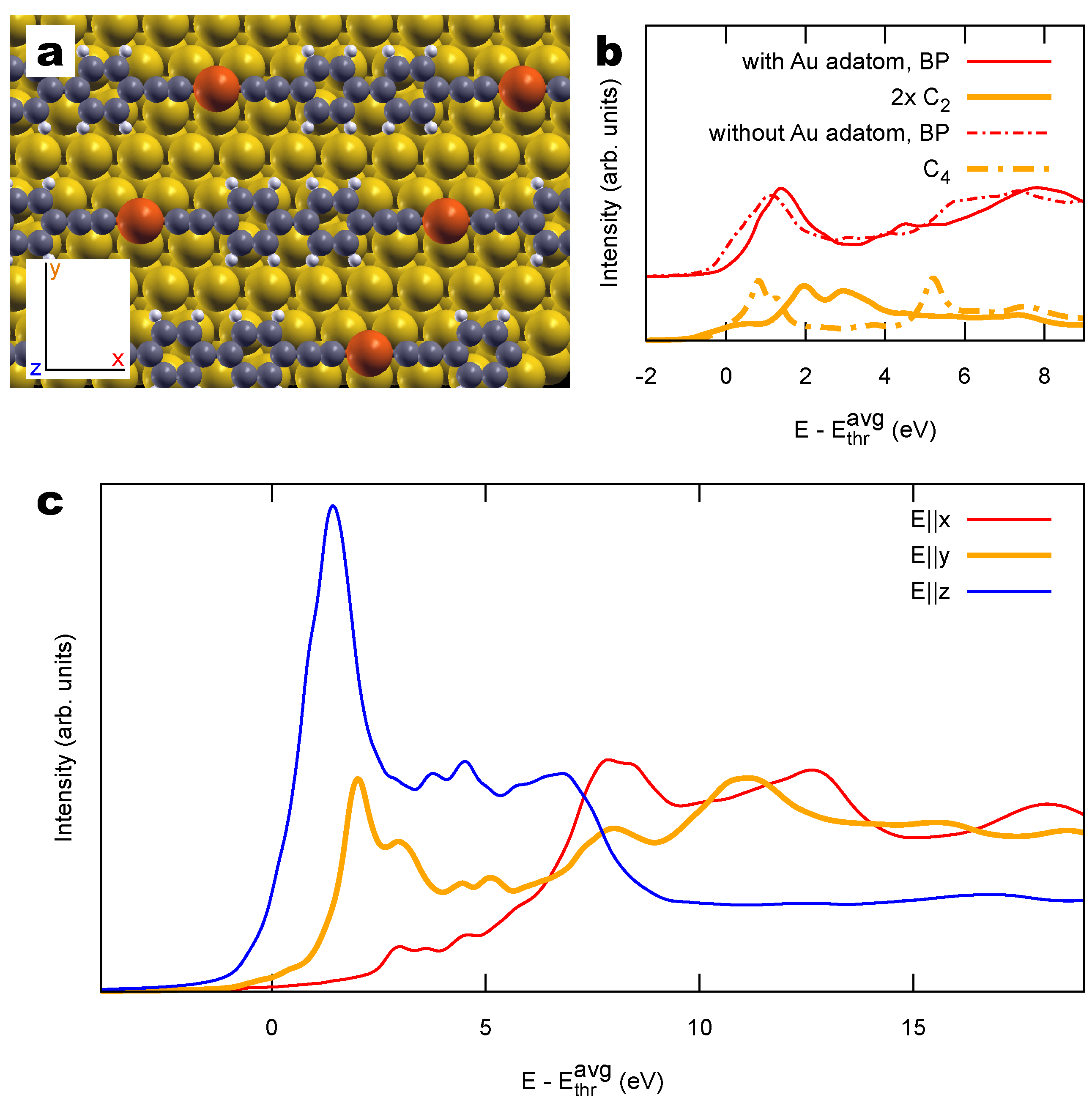
2.2. Combined sp/sp Polymers on a Metal Surface
3. Discussion
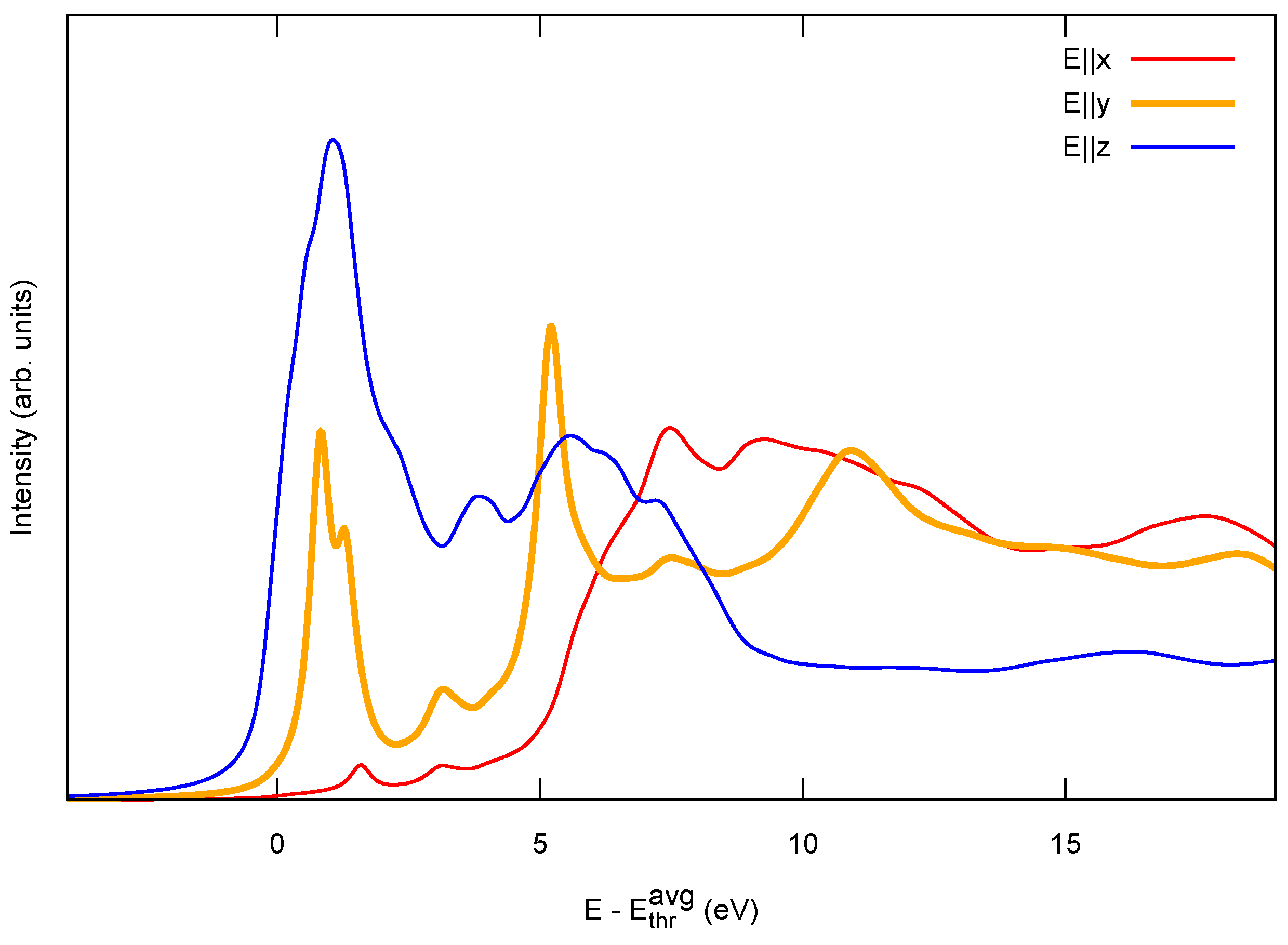
3.1. Identification of sp C Chains in NEXAFS Spectra
3.2. Other Structural Models
4. Materials and Methods
4.1. Theoretical Framework
4.2. Numerical Protocol
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1D | one-dimensional |
| 2D | two-dimensional |
| AFM | atomic force microscopy |
| bBEBP | 4,4-di(bromoethynyl)-1,1-biphenyl |
| BLA | bond length alternation |
| BP | biphenyl |
| DOS | density of states |
| DFT | density functional theory |
| FCC | face-centered cubic |
| HCP | hexagonal close packed |
| KPDOS | k-resolved projected density of states |
| NEXAFS | near-edge X-ray absorption fine structure |
| PBE | Perdew, Burke, and Ernzerhof |
| PDOS | projected density of states |
| PPP | poly(p-phenylene) |
| STM | scanning tunneling microscopy |
| tBEB | 1,3,5-tris(bromoethynyl)benzene |
| UHV | ultra-high vacuum |
| XPS | X-ray photoemission spectroscopy |
References
- Hirsch, A. The era of Carbon allotropes. Nat. Mater. 2010, 9, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Castelli, I.E.; Salvestrini, P.; Manini, N. Mechanical properties of carbynes investigated by ab-initio total-energy calculations. Phys. Rev. B 2012, 85, 214110. [Google Scholar] [CrossRef]
- Hoffmann, R.; Kabanov, A.A.; Golov, A.A.; Proserpio, D.M. Homo Citans and Carbon Allotropes: For an Ethics of Citation. Angew. Chem. Int. Ed. 2016, 55, 10962. [Google Scholar] [CrossRef] [PubMed]
- Burchfield, L.; Fahim, M.A.; Wittman, R.; Delodovici, F.; Manini, N. Novamene: A new class of carbon allotropes. Heliyon 2017, 3, e00242. [Google Scholar] [CrossRef] [PubMed]
- Delodovici, F.; Manini, N.; Wittman, R.; Choi, D.; Fahim, M.A.; Burchfield, L. Protomene: A new carbon allotrope. Carbon 2018, 126, 574. [Google Scholar] [CrossRef]
- Casari, C.S.; Tommasini, M.; Tykwinski, R.R.; Milani, A. Carbon-atom wires: 1-D systems with tunable properties. Nanoscale 2016, 8, 4414–4435. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, F. (Ed.) Polyynes: Synthesis, Properties, and Applications; CRC, Taylor & Francis: London, UK, 2005. [Google Scholar]
- Casari, C.S.; Milani, A. Carbyne: From the elusive allotrope to stable Carbon atom wires. MRS Commun. 2018, 8, 207–219. [Google Scholar] [CrossRef]
- Banhart, F. Chains of Carbon atoms: A vision or a new nanomaterial? Beilstein J. Nanotechnol. 2015, 6, 559–569. [Google Scholar] [CrossRef]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of polyynes to model the sp-Carbon allotrope carbyne. Nat. Chem. 2010, 2, 967–971. [Google Scholar] [CrossRef]
- Shi, L.; Rohringer, P.; Suenaga, K.; Niimi, Y.; Kotakoski, J.; Meyer, J.C.; Peterlik, H.; Wanko, M.; Cahangirov, S.; Rubio, A.; et al. Confined linear Carbon chains as a route to bulk carbyne. Nat. Mater. 2016, 15, 634. [Google Scholar] [CrossRef]
- Movsisyan, L.D.; Kondratuk, D.V.; Franz, M.; Thompson, A.L.; Tykwinski, R.R.; Anderson, H.L. Synthesis of Polyyne Rotaxanes. Org. Lett. 2012, 14, 3424–3426. [Google Scholar] [CrossRef] [PubMed]
- Movsisyan, L.D.; Franz, M.; Hampel, F.; Thompson, A.L.; Tykwinski, R.R.; Anderson, H.L. Polyyne Rotaxanes: Stabilization by Encapsulation. J. Am. Chem. Soc. 2016, 138, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Januszewski, J.A.; Wendinger, D.; Neiss, C.; Movsisyan, L.D.; Hampel, F.; Anderson, H.L.; Goerling, A.; Tykwinski, R.R. Cumulene Rotaxanes: Stabilization and Study of [9]Cumulenes. Angew. Chem. Int. Ed. 2015, 54, 6645–6649. [Google Scholar] [CrossRef] [PubMed]
- Weisbach, N.; Baranova, Z.; Gauthier, S.; Reibenspies, J.H.; Gladysz, J.A. A new type of insulated molecular wire: A rotaxane derived from a metal-capped conjugated tetrayne. Chem. Commun. 2012, 48, 7562–7564. [Google Scholar] [CrossRef] [PubMed]
- Schrettl, S.; Contal, E.; Hoheisel, T.N.; Fritzsche, M.; Balog, S.; Szilluweit, R.; Frauenrath, H. Facile synthesis of oligoyne amphiphiles and their rotaxanes. Chem. Sci. 2015, 6, 564–574. [Google Scholar] [CrossRef]
- Malko, D.; Neiss, C.; Vines, F.; Goerling, A. Competition for Graphene: Graphynes with Direction-Dependent Dirac Cones. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef] [PubMed]
- Castelli, I.E.; Ferri, N.; Onida, G.; Manini, N. Carbon sp chains in graphene nanoholes. J. Phys. Condens. Matter 2012, 24, 104019. [Google Scholar] [CrossRef]
- Ivanovskii, A.L. Graphynes and graphdyines. Prog. Solid State Chem. 2013, 41, 1–19. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Liu, H.; Li, Y. Graphdiyne and graphyne: From theoretical predictions to practical construction. Chem. Soc. Rev. 2014, 43, 2572–2586. [Google Scholar] [CrossRef]
- Diederich, F.; Kivala, M. All-Carbon Scaffolds by Rational Design. Adv. Mater. 2010, 22, 803–812. [Google Scholar] [CrossRef]
- Ravagnan, L.; Siviero, F.; Lenardi, C.; Piseri, P.; Barborini, E.; Milani, P.; Casari, C.; Li Bassi, A.; Bottani, C. Cluster-beam deposition and in situ characterization of carbyne-rich Carbon films. Phys. Rev. Lett. 2002, 89. [Google Scholar] [CrossRef]
- Casari, C.S.; Giannuzzi, C.S.; Russo, V. Carbon-atom wires produced by nanosecond pulsed laser deposition in a background gas. Carbon 2016, 104, 190–195. [Google Scholar] [CrossRef]
- Cataldo, F. Synthesis of polyynes in a submerged electric arc in organic solvents. Carbon 2004, 42, 129–142. [Google Scholar] [CrossRef]
- Tabata, H.; Fujii, M.; Hayashi, S.; Doi, T.; Wakabayashi, T. Raman and surface-enhanced Raman scattering of a series of size-separated polyynes. Carbon 2006, 44, 3168–3176. [Google Scholar] [CrossRef]
- Szafert, S.; Gladysz, J.A. Update 1 of: Carbon in one dimension: Structural analysis of the higher conjugated polyynes. Chem. Rev. 2006, 106, PR1–PR33. [Google Scholar] [CrossRef]
- Tykwinski, R.R.; Chalifoux, W.; Eisler, S.; Lucotti, A.; Tommasini, M.; Fazzi, D.; Del Zoppo, M.; Zerbi, G. Toward carbyne: Synthesis and stability of really long polyynes. Pure Appl. Chem. 2010, 82, 891–904. [Google Scholar] [CrossRef]
- Cataldo, F.; Ursini, O.; Milani, A.; Casari, C.S. One-pot synthesis and characterization of polyynes end-capped by biphenyl groups (α, ω-biphenylpolyynes). Carbon 2018, 126, 232–240. [Google Scholar] [CrossRef]
- Cataldo, F.; Ursini, O.; Angelini, G.; Tommasini, M.; Casari, C. Simple Synthesis of, -Diarylpolyynes Part 1: Diphenylpolyynes. J. Marcomol. Sci. A 2010, 47, 739–746. [Google Scholar] [CrossRef]
- Sun, Q.; Cai, L.; Ma, H.; Yuan, C.; Xu, W. Dehalogenative Homocoupling of Terminal Alkynyl Bromides on Au(111): Incorporation of Acetylenic Scaffolding into Surface Nanostructures. ACS Nano 2016, 10, 7023–7030. [Google Scholar] [CrossRef]
- Sun, Q.; Tran, B.V.; Cai, L.; Ma, H.; Yu, X.; Yuan, C.; Stohr, M.; Xu, W. On-Surface Formation of Cumulene by Dehalogenative Homocoupling of Alkenyl gem-Dibromides. Angw. Chem. Int. Ed. 2017, 56, 12165–12169. [Google Scholar] [CrossRef]
- Shu, C.H.; Liu, M.X.; Zha, Z.Q.; Pan, J.L.; Zhang, S.Z.; Xie, Y.L.; Chen, J.L.; Yuan, D.W.; Qiu, X.H.; Liu, P.N. On-surface synthesis of poly(p-phenylene ethynylene) molecular wires via in situ formation of Carbon-Carbon triple bond. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Griesing, S.; Rybachuk, M.; Lu, Q.B.; Duley, W.W. Nanobuckling and X-ray photoelectron spectra of carbyne-rich tetrahedral Carbon films deposited by femtosecond laser ablation at cryogenic temperatures. J. Appl. Phys. 2007, 102. [Google Scholar] [CrossRef]
- Ravagnan, L.; Bongiorno, G.; Bandiera, D.; Salis, E.; Piseri, P.; Milani, P.; Lenardi, C.; Coreno, M.; de Simone, M.; Prince, K.C. Quantitative evaluation of sp/sp2 hybridization ratio in cluster-assembled Carbon films by in situ near edge X-ray absorption fine structure spectroscopy. Carbon 2006, 44, 1518–1524. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy; Springer: Berlin/Heidelberg, Germany, 1992; Volume 25. [Google Scholar]
- Hähner, G. Near edge X-ray absorption fine structure spectroscopy as a tool to probe electronic and structural properties of thin organic films and liquids. Chem. Soc. Rev. 2006, 35, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Mazza, T.; Bongiorno, G.; Devetta, M.; Amati, M.; Milani, P.; Piseri, P.; Coreno, M.; Lenardi, C.; Evangelista, F.; et al. sp hybridization in free carbon nanoparticles—Presence and stability observed by near edge X-ray absorption fine structure spectroscopy. Chem. Commun. 2011, 47, 2952–2954. [Google Scholar] [CrossRef] [PubMed]
- Haerle, R.; Riedo, E.; Pasquarello, A.; Baldereschi, A. sp2/sp3 hybridization ratio in amorphous carbon from C 1s core-level shifts: X-ray photoelectron spectroscopy and first-principles calculation. Phys. Rev. B 2001, 65, 045101. [Google Scholar] [CrossRef]
- Titantah, J.; Lamoen, D. sp3/sp2 characterization of carbon materials from first-principles calculations: X-ray photoelectron versus high energy electron energy-loss spectroscopy techniques. Carbon 2005, 43, 1311–1316. [Google Scholar] [CrossRef]
- Levi, G.; Senneca, O.; Causà, M.; Salatino, P.; Lacovig, P.; Lizzit, S. Probing the chemical nature of surface oxides during coal char oxidation by high-resolution XPS. Carbon 2015, 90, 181–196. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Gilmore, K.; Vinson, J.; Shirley, E.; Prendergast, D.; Pemmaraju, C.; Kas, J.; Vila, F.; Rehr, J. Efficient implementation of core-excitation Bethe–Salpeter equation calculations. Comput. Phys. Commun. 2015, 197, 109–117. [Google Scholar] [CrossRef]
- Slater, J.C.; Johnson, K.H. Self-Consistent-Field Xα Cluster Method for Polyatomic Molecules and Solids. Phys. Rev. B 1972, 5, 844–853. [Google Scholar] [CrossRef]
- Triguero, L.; Pettersson, L.G.M.; Ågren, H. Calculations of near-edge X-ray-absorption spectra of gas-phase and chemisorbed molecules by means of density-functional and transition-potential theory. Phys. Rev. B 1998, 58, 8097–8110. [Google Scholar] [CrossRef]
- Leetmaa, M.; Ljungberg, M.P.; Lyubartsev, A.; Nilsson, A.; Pettersson, L.G.M. Theoretical approximations to X-ray absorption spectroscopy of liquid water and ice. J. Electron. Spectrosc. Relat. Phenom. 2010, 177, 135–157. [Google Scholar] [CrossRef]
- Ugolotti, A.; Harivyasi, S.S.; Baby, A.; Dominguez, M.; Pinardi, A.L.; López, M.F.; Martín-Gago, J.Á.; Fratesi, G.; Floreano, L.; Brivio, G.P. Chemisorption of Pentacene on Pt(111) with a Little Molecular Distortion. J. Phys. Chem. C 2017, 121, 22797–22805. [Google Scholar] [CrossRef]
- Perera, S.D.; Urquhart, S.G. Systematic Investigation of π–π Interactions in Near-Edge X-ray Fine Structure (NEXAFS) Spectroscopy of Paracyclophanes. J. Phys. Chem. A 2017, 121, 4907–4913. [Google Scholar] [CrossRef] [PubMed]
- Diller, K.; Maurer, R.J.; Müller, M.; Reuter, K. Interpretation of X-ray absorption spectroscopy in the presence of surface hybridization. J. Chem. Phys. 2017, 146, 214701. [Google Scholar] [CrossRef]
- Baby, A.; Fratesi, G.; Vaidya, S.R.; Patera, L.L.; Africh, C.; Floreano, L.; Brivio, G.P. Anchoring and Bending of Pentacene on Aluminum (001). J. Phys. Chem. C 2015, 119, 3624–3633. [Google Scholar] [CrossRef]
- Baby, A.; Lin, H.; Ravikumar, A.; Bittencourt, C.; Wegner, H.A.; Floreano, L.; Goldoni, A.; Fratesi, G. Lattice Mismatch Drives Spatial Modulation of Corannulene Tilt on Ag(111). J. Phys. Chem. C 2018, 122, 10365–10376. [Google Scholar] [CrossRef]
- Fratesi, G.; Lanzilotto, V.; Floreano, L.; Brivio, G.P. Azimuthal Dichroism in Near-Edge X-ray Absorption Fine Structure Spectra of Planar Molecules. J. Phys. Chem. C 2013, 117, 6632–6638. [Google Scholar] [CrossRef]
- Fratesi, G.; Lanzilotto, V.; Stranges, S.; Alagia, M.; Brivio, G.P.; Floreano, L. High resolution NEXAFS of perylene and PTCDI: A surface science approach to molecular orbital analysis. Phys. Chem. Chem. Phys. 2014, 16, 14834–14844. [Google Scholar] [CrossRef] [PubMed]
- Heimann, R.; Kleiman, J.; Salansky, N. A unified structural approach to linear carbon polytypes. Nature 1983, 306, 164–167. [Google Scholar] [CrossRef]
- Milani, A.; Tommasini, M.; Del Zoppo, M.; Castiglioni, C.; Zerbi, G. Carbon nanowires: Phonon and π-electron confinement. Phys. Rev. B 2006, 74. [Google Scholar] [CrossRef]
- Stöhr, J.; Sette, F.; Johnson, A.L. Near-Edge X-Ray-Absorption Fine-Structure Studies of Chemisorbed Hydrocarbons: Bond Lengths with a Ruler. Phys. Rev. Lett. 1984, 53, 1684–1687. [Google Scholar] [CrossRef]
- Kovacic, P.; Kyriakis, A. Polymerization of benzene to p-polyphenyl. Tetrahedron Lett. 1962, 3, 467–469. [Google Scholar] [CrossRef]
- Grein, F. Twist Angles and Rotational Energy Barriers of Biphenyl and Substituted Biphenyls. J. Phys. Chem. A 2002, 106, 3823–3827. [Google Scholar] [CrossRef]
- Donati, F.; Fratesi, G.; Passoni, M.; Casari, C.S.; Mairov, A.; Bottani, C.E.; Trioni, M.I.; Li Bassi, A. Strain effect on local electronic properties of Fe nanoislands grown on Au(111). Phys. Rev. B 2011, 83, 153404. [Google Scholar] [CrossRef]
- Sirotti, F.; Beaulieu, N.; Bendounan, A.; Silly, M.G.; Chauvet, C.; Malinowski, G.; Fratesi, G.; Véniard, V.; Onida, G. Multiphoton k-resolved photoemission from gold surface states with 800-nm femtosecond laser pulses. Phys. Rev. B 2014, 90, 035401. [Google Scholar] [CrossRef]
- Achilli, S.; Caravati, S.; Trioni, M.I. Ab initio electronic and magnetic properties of 1 ML Fe/Cu(001). J. Phys. Condens. Matter 2007, 19, 305021. [Google Scholar] [CrossRef]
- Brivio, G.P.; Butti, G.; Caravati, S.; Fratesi, G.; Trioni, M.I. Theoretical approaches in adsorption: Alkali adatom investigations. J. Phys. Condens. Matter 2007, 19, 305005. [Google Scholar] [CrossRef]
- Klappenberger, F.; Zhang, Y.Q.; Björk, J.; Klyatskaya, S.; Ruben, M.; Barth, J.V. On-Surface Synthesis of Carbon-Based Scaffolds and Nanomaterials Using Terminal Alkynes. Accounts Chem. Res. 2015, 48, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Fratesi, G. First-principles investigation of the early stages of Pd adsorption on Au(111). J. Phys. Condens. Matter 2011, 23, 015001. [Google Scholar] [CrossRef] [PubMed]
- Gougoussis, C.; Calandra, M.; Seitsonen, A.P.; Mauri, F. First-principles calculations of X-ray absorption in a scheme based on ultrasoft pseudopotentials: From α-quartz to high-Tc compounds. Phys. Rev. B 2009, 80, 075102. [Google Scholar] [CrossRef]
- Pehlke, E.; Scheffler, M. Evidence for site-sensitive screening of core holes at the Si and Ge(001) surface. Phys. Rev. Lett. 1993, 71, 2338–2341. [Google Scholar] [CrossRef]
- Liu, M.; Artyukhov, V.I.; Lee, H.; Xu, F.; Yakobson, B.I. Carbyne from first principles: Chain of C atoms, a nanorod or a nanorope. ACS Nano 2013, 7, 10075. [Google Scholar] [CrossRef]
- Tumino, F.; Rabia, A.; Russo, V.; Milani, A.; Bassi, A.L.; Achilli, S.; Fratesi, G.; Onida, G.; Manini, N.; Trioni, M.I.; et al. Atomic scale structure, electronic and vibrational properties of one-dimensional sp-sp2 carbon-based nanostructures. in preparation.
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fratesi, G.; Achilli, S.; Manini, N.; Onida, G.; Baby, A.; Ravikumar, A.; Ugolotti, A.; Brivio, G.P.; Milani, A.; Casari, C.S. Fingerprints of sp1 Hybridized C in the Near-Edge X-ray Absorption Spectra of Surface-Grown Materials. Materials 2018, 11, 2556. https://doi.org/10.3390/ma11122556
Fratesi G, Achilli S, Manini N, Onida G, Baby A, Ravikumar A, Ugolotti A, Brivio GP, Milani A, Casari CS. Fingerprints of sp1 Hybridized C in the Near-Edge X-ray Absorption Spectra of Surface-Grown Materials. Materials. 2018; 11(12):2556. https://doi.org/10.3390/ma11122556
Chicago/Turabian StyleFratesi, Guido, Simona Achilli, Nicola Manini, Giovanni Onida, Anu Baby, Abhilash Ravikumar, Aldo Ugolotti, Gian Paolo Brivio, Alberto Milani, and Carlo Spartaco Casari. 2018. "Fingerprints of sp1 Hybridized C in the Near-Edge X-ray Absorption Spectra of Surface-Grown Materials" Materials 11, no. 12: 2556. https://doi.org/10.3390/ma11122556
APA StyleFratesi, G., Achilli, S., Manini, N., Onida, G., Baby, A., Ravikumar, A., Ugolotti, A., Brivio, G. P., Milani, A., & Casari, C. S. (2018). Fingerprints of sp1 Hybridized C in the Near-Edge X-ray Absorption Spectra of Surface-Grown Materials. Materials, 11(12), 2556. https://doi.org/10.3390/ma11122556