Influence of Oxygen Vacancy Density on the Polaronic Configuration in Rutile



Abstract
1. Introduction
2. Computational Methods
- Calculate linear response on single Ti atom in perfect rutile crystal without setting any U value. A value of = 4.2 eV can be obtained for Ti-3d orbitals.
- Calculate band gap with = 4.2 eV and variable . A value of = 4.5 eV fits the experimental value.
- Re-calculate linear response of single Ti atom in perfect rutile crystal with = 4.5 eV. A new value of about = 4.0 eV can be obtained with this . The use of will lower electron population on Ti atoms, which decrease the linear response result from = 4.2 eV to 4.0 eV.
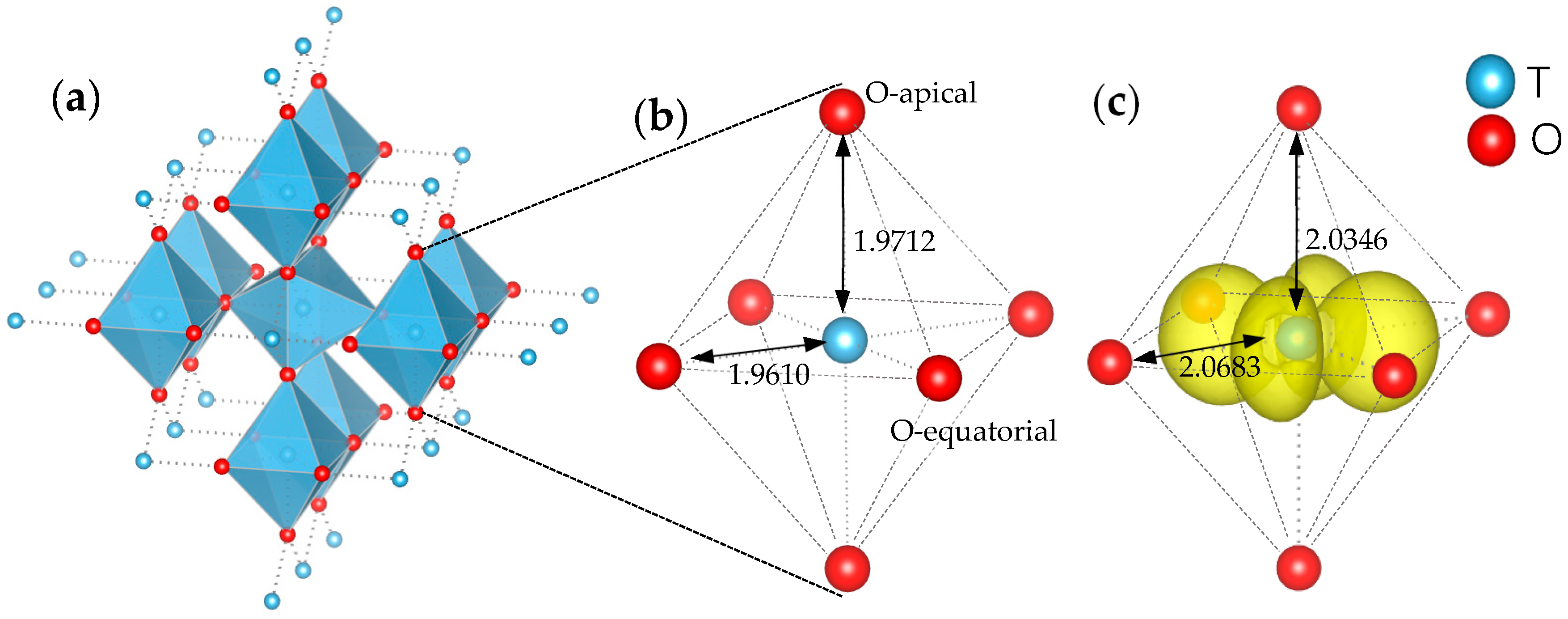
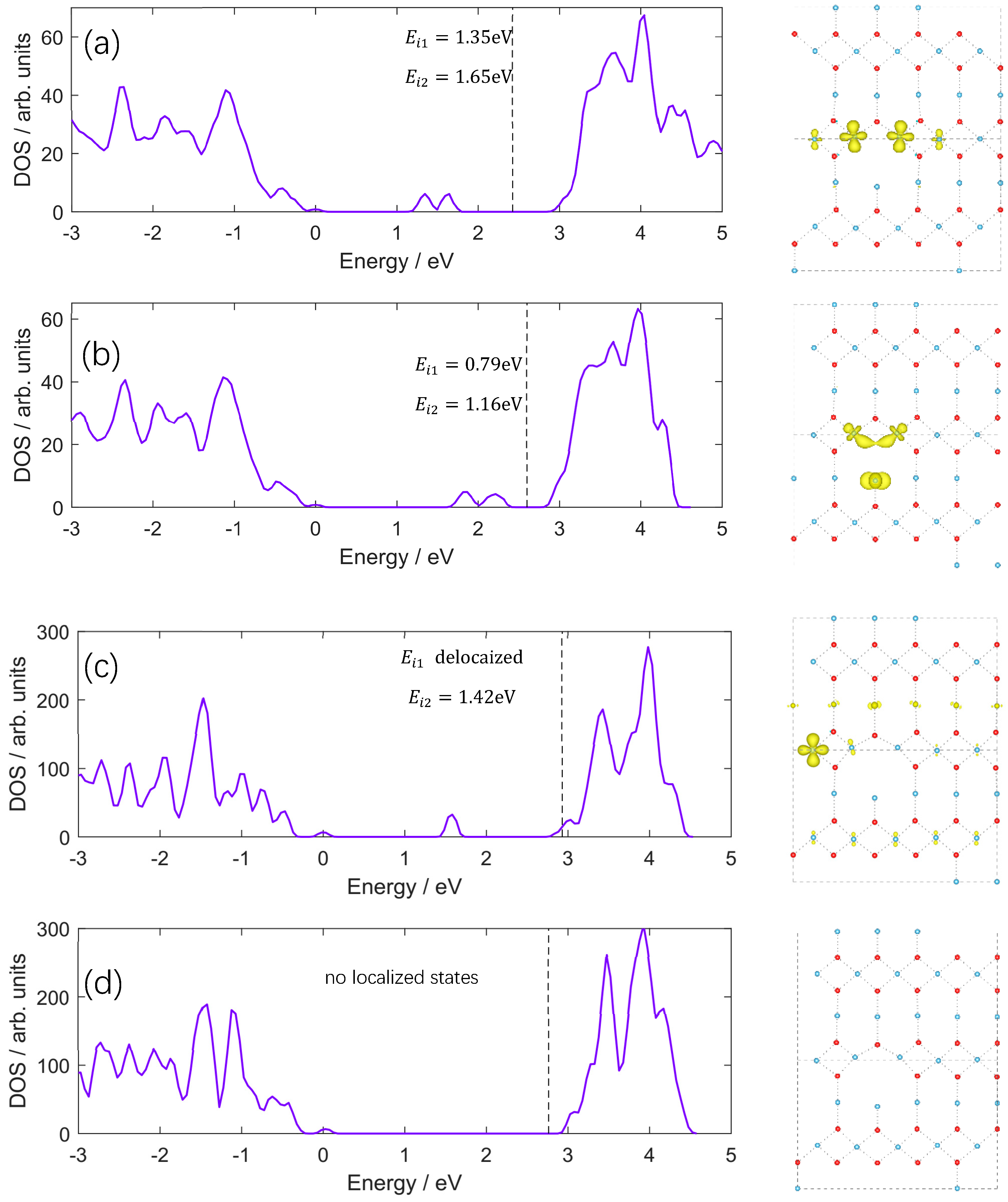
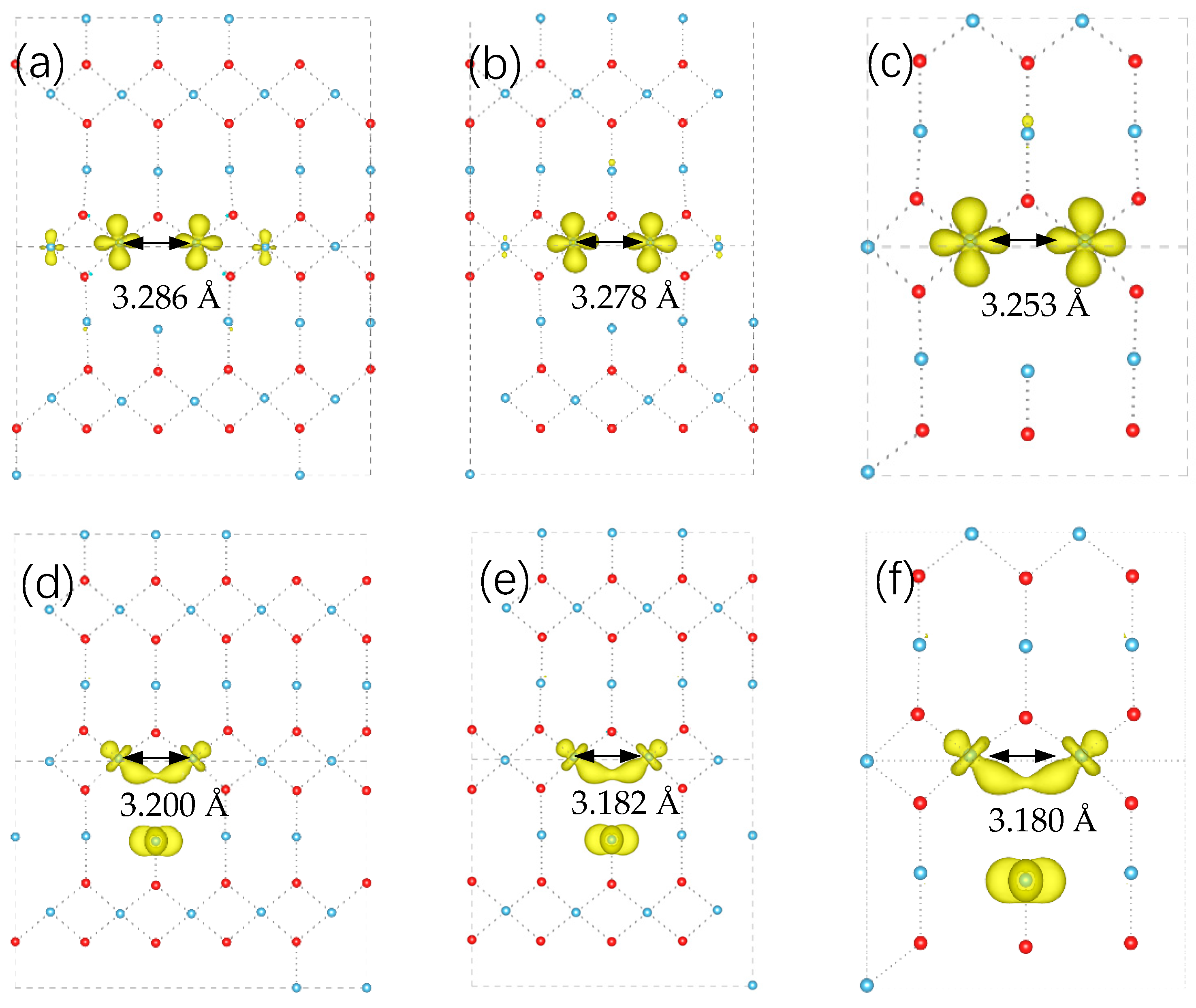
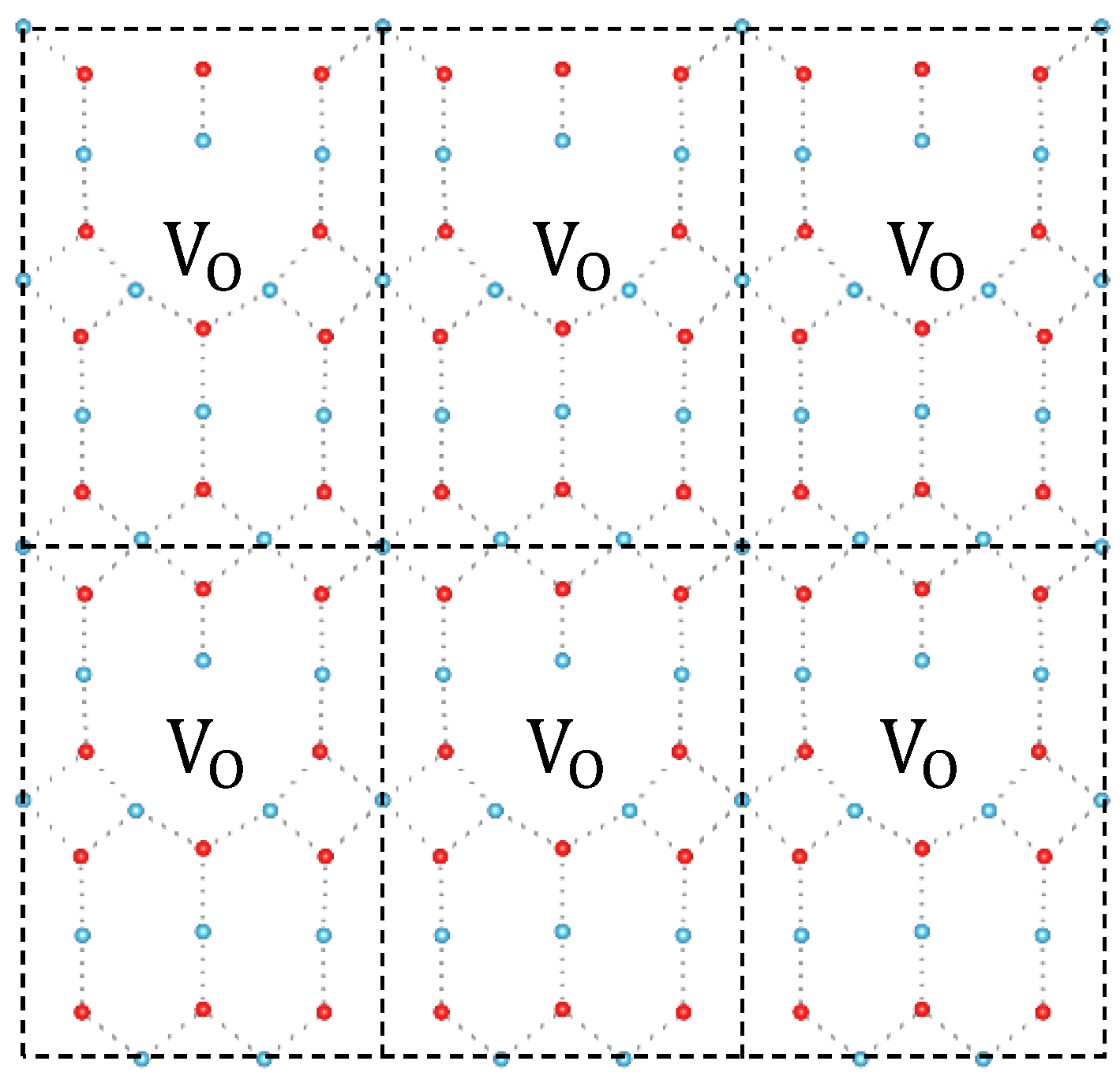
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Nowotny, J. Oxide Semiconductors for Solar Energy Conversion—Titanium Dioxide; CRC Press: New York, NY, USA, 2012; p. 150. [Google Scholar]
- Tang, Z.; Fang, L.; Xu, N.; Liu, R. Forming compliance dominated memristive switching through interfacial reaction in Ti/TiO2/Au structure. J. Appl. Phys. 2015, 118, 185309. [Google Scholar] [CrossRef]
- Tang, Z.; Chi, Y.; Fang, L.; Liu, R.; Yi, X. Resistive switching effect in titanium oxides. J. Nanosci. Nanotechnol. 2014, 14, 1494–1507. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.G.; Flavell, W.R.; Mallick, A.K.; Kumarasinghe, A.R.; Tsoutsou, D.; Khan, N.; Chatwin, C.; Rayner, S.; Smith, G.C.; Stockbauer, R.L.; et al. Comparison of the electronic structure of anatase and rutile TiO2 single-crystal surfaces using resonant photoemission and x-ray absorption spectroscopy. Phys. Rev. B 2007, 75, 035105. [Google Scholar] [CrossRef]
- Thomas, A.G.; Flavell, W.R.; Kumarasinghe, A.R.; Mallick, A.K.; Tsoutsou, D.; Smith, G.C.; Stockbauer, R.; Patel, S.; Grätzel, M.; Hengerer, R. Resonant photoemission of anatase TiO2 (101) and (001) single crystals. Phys. Rev. B 2003, 67, 035110. [Google Scholar] [CrossRef]
- Cronemeyer, D.C. Infrared Absorption of Reduced Rutile TiO2 Single Crystals. Phys. Rev. 1959, 113, 1222. [Google Scholar] [CrossRef]
- Lin, C.; Shin, D.; Demkov, A.A. Localized states induced by an oxygen vacancy in rutile TiO2. J. Appl. Phys. 2015, 117, 225703. [Google Scholar] [CrossRef]
- Deák, P.; Aradi, B.; Frauenheim, T. Quantitative theory of the oxygen vacancy and carrier self-trapping in bulk TiO2. Phys. Rev. B 2012, 86, 195206. [Google Scholar] [CrossRef]
- Janotti, A.; Varley, J.B.; Rinke, P.; Umezawa, N.; Kresse, G.; Van de Walle, C.G. Hybrid functional studies of the oxygen vacancy in TiO2. Phys. Rev. B 2010, 81, 085212. [Google Scholar] [CrossRef]
- Vásquez, G.C.; Karazhanov, S.Z.; Maestre, D.; Cremades, A.; Piqueras, J.; Foss, S.E. Oxygen vacancy related distortions in rutile TiO2 nanoparticles: A combined experimental and theoretical study. Phys. Rev. B 2016, 94, 235209. [Google Scholar] [CrossRef]
- Deák, P.; Aradi, B.; Frauenheim, T. Oxygen deficiency in TiO2: Similarities and differences between the Ti self-interstitial and the O vacancy in bulk rutile and anatase. Phys. Rev. B 2015, 92, 045204. [Google Scholar] [CrossRef]
- Deák, P.; Kullgren, J.; Frauenheim, T. Polarons and oxygen vacancies at the surface of anatase TiO2. Phys. Status Solid RRL 2014, 8, 583–586. [Google Scholar] [CrossRef]
- Deák, P.; Aradi, B.; Frauenheim, T. Polaronic effects in TiO2 calculated by the HSE06 hybrid functional: Dopant passivation by carrier self-trapping. Phys. Rev. B 2011, 83, 155207. [Google Scholar] [CrossRef]
- Morgan, B.J.; Watson, G.W. Intrinsic n-type Defect Formation in TiO2: A Comparison of Rutile and Anatase from GGA + U Calculations. J. Phys. Chem. C 2010, 114, 2321–2328. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B.; Feng, Y.P. Density-functional characterization of antiferromagnetism in oxygen-deficient anatase and rutile TiO2. Phys. Rev. B 2010, 81, 033202. [Google Scholar] [CrossRef]
- Finazzi, E.; Di Valentin, C.; Pacchioni, G.; Selloni, A. Excess electron states in reduced bulk anatase TiO2: Comparison of standard GGA, GGA + U, and hybrid DFT calculations. J. Chem. Phys. 2008, 129, 154113. [Google Scholar] [CrossRef] [PubMed]
- Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Localization and Delocalization Errors in Density Functional Theory and Implications for Band-Gap Prediction. Phys. Rev. Lett. 2008, 100, 146401. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.J.; Watson, G.W. Polaronic trapping of electrons and holes by native defects in anatase TiO2. Phys. Rev. B 2009, 80, 233102. [Google Scholar] [CrossRef]
- Park, S.G.; Magyari-Köpe, B.; Nishi, Y. Electronic correlation effects in reduced rutile TiO2 within the LDA + U method. Phys. Rev. B 2010, 82, 1053. [Google Scholar] [CrossRef]
- Tilocca, A.; Selloni, A. DFT-GGA and DFT + U Simulations of Thin Water Layers on Reduced TiO2 Anatase. J. Phys. Chem. C 2012, 116, 9114–9121. [Google Scholar] [CrossRef]
- Stausholmmøller, J.; Kristoffersen, H.H.; Hinnemann, B.; Madsen, G.K.; Hammer, B. DFT + U study of defects in bulk rutile TiO2. J. Chem. Phys. 2010, 133, 144708. [Google Scholar] [CrossRef] [PubMed]
- Shao, G. Red Shift in Manganese- and Iron-Doped TiO2: A DFT + U Analysis. J. Phys. Chem. C 2009, 113, 6800–6808. [Google Scholar] [CrossRef]
- Valentin, C.D.; Pacchioni, G.; Selloni, A. Reduced and n-Type Doped TiO2: Nature of Ti3+ Species. J. Phys. Chem. C 2009, 113, 20543–20552. [Google Scholar] [CrossRef]
- Islam, M.M.; Bredow, T.; Gerson, A. Electronic properties of oxygen-deficient and aluminum-doped rutile TiO2 from first principles. Phys. Rev. B 2007, 76, 045217. [Google Scholar] [CrossRef]
- Finazzi, E.; Di Valentin, C.; Pacchioni, G. Nature of Ti Interstitials in Reduced Bulk Anatase and Rutile TiO2. J. Phys. Chem. C 2009, 113, 3382–3385. [Google Scholar] [CrossRef]
- Lechermann, F.; Heckel, W.; Kristanovski, O.; Müller, S. Oxygen-vacancy driven electron localization and itinerancy in rutile-based TiO2. Phys. Rev. B 2017, 95, 195159. [Google Scholar] [CrossRef]
- Malashevich, A.; Jain, M.; Louie, S.G. First-principles DFT + GW study of oxygen vacancies in rutile TiO2. Phys. Rev. B 2014, 89, 075205. [Google Scholar] [CrossRef]
- Hubbard, J. Electron Correlations in Narrow Energy Bands. Proc. R. Soc. Lond. A 1963, 276, 238–257. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I. Quantum Espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Mater. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Constantin, L.A.; Terentjevs, A.; Della Sala, F.; Fabiano, E. Gradient-dependent upper bound for the exchange-correlation energy and application to density functional theory. Phys. Rev. B 2015, 91, 041120. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2007, 101, 136406. [Google Scholar] [CrossRef] [PubMed]
- Standard Solid State Pseudopotentials (SSSP). Available online: http://materialscloud.org/sssp/ (accessed on 30 October 2018).
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal, C.A. Reproducibility in density functional theory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef] [PubMed]
- Kulik, H.J.; Matteo, C.; Scherlis, D.A.; Nicola, M. Density functional theory in transition-metal chemistry: A self-consistent Hubbard U approach. Phys. Rev. Lett. 2006, 97, 103001. [Google Scholar] [CrossRef] [PubMed]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA + U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef]
- Hybertsen, M.S.; Schlüter, M.; Christensen, N.E. Calculation of Coulomb-interaction parameters for La2CuO4 using a constrained-density-functional approach. Phys. Rev. B 1989, 39, 9028. [Google Scholar] [CrossRef]
- Lany, S.; Zunger, A. Polaronic hole localization and multiple hole binding of acceptors in oxide wide-gap semiconductors. Phys. Rev. B 2009, 80, 085202. [Google Scholar] [CrossRef]
- McMahan, A.K.; Martin, R.M.; Satpathy, S. Calculated effective Hamiltonian for La2CuO4 and solution in the impurity Anderson approximation. Phys. Rev. B 1988, 38, 6650. [Google Scholar] [CrossRef]
- Troubleshooting Common Problems with DFT + U. Available online: http://hjkgrp.mit.edu/content/troubleshooting-common-problems-dftu (accessed on 24 October 2018).
- Pascual, J.; Camassel, J.; Mathieu, H. Fine structure in the intrinsic absorption edge of TiO2. Phys. Rev. B 1978, 18, 5606. [Google Scholar] [CrossRef]
- Pascual, J.; Camassel, J.; Mathieu, H. Resolved Quadrupolar Transition in TiO2. Phys. Rev. Lett. 1977, 39, 1490. [Google Scholar] [CrossRef]
- Handbook of Mineralogy. Available online: http://www.handbookofmineralogy.org/pdfs/rutile.pdf (accessed on 30 October 2018).
- Asaduzzaman, A.M.; Krüger, P. A First Principles Study on Charge Dependent Diffusion of Point Defects in Rutile TiO2. J. Phys. Chem. C 2010, 114, 10649. [Google Scholar] [CrossRef]
- Yagi, E.; Hasiguti, R.R.; Aono, M. Electronic conduction above 4 K of slightly reduced oxygen-deficient rutile TiO2−x. Phys. Rev. B 1996, 54, 7945. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Normal Ti | Equatorial Ti | Apical Ti |
|---|---|---|---|
| (a) | 2.50 | 2.15 (0.35) | 2.42 (0.08) |
| (b) | 2.50 | 2.29 (0.21) | 2.12 (0.38) |
| Index | U of Equatorial Ti | U of Apical Ti | Index | U of Equatorial Ti | U of Apical Ti |
|---|---|---|---|---|---|
| (a) | 4.81 eV | 4.00 eV | (d) | 4.66 eV | 5.18 eV |
| (b) | 4.80 eV | 4.01 eV | (e) | 4.01 eV | 5.18 eV |
| (c) | 5.03 eV | 4.12 eV | (f) | 3.87 eV | 5.32 eV |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Fang, L.; Hao, Y.; Chi, Y. Influence of Oxygen Vacancy Density on the Polaronic Configuration in Rutile. Materials 2018, 11, 2156. https://doi.org/10.3390/ma11112156
Liu R, Fang L, Hao Y, Chi Y. Influence of Oxygen Vacancy Density on the Polaronic Configuration in Rutile. Materials. 2018; 11(11):2156. https://doi.org/10.3390/ma11112156
Chicago/Turabian StyleLiu, Rulin, Liang Fang, Yue Hao, and Yaqing Chi. 2018. "Influence of Oxygen Vacancy Density on the Polaronic Configuration in Rutile" Materials 11, no. 11: 2156. https://doi.org/10.3390/ma11112156
APA StyleLiu, R., Fang, L., Hao, Y., & Chi, Y. (2018). Influence of Oxygen Vacancy Density on the Polaronic Configuration in Rutile. Materials, 11(11), 2156. https://doi.org/10.3390/ma11112156