Electrochemically Treated TiO2 for Enhanced Performance in Aqueous Al-Ion Batteries




Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Electrode Characterisation
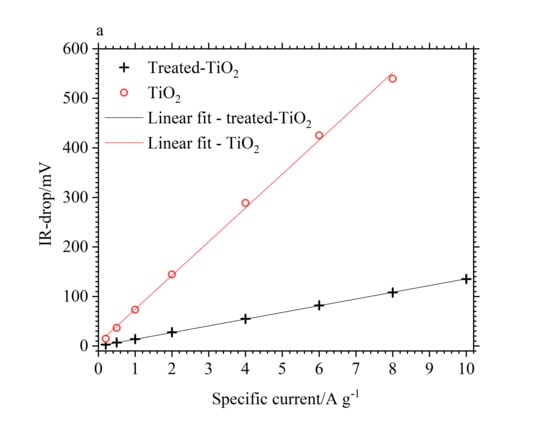
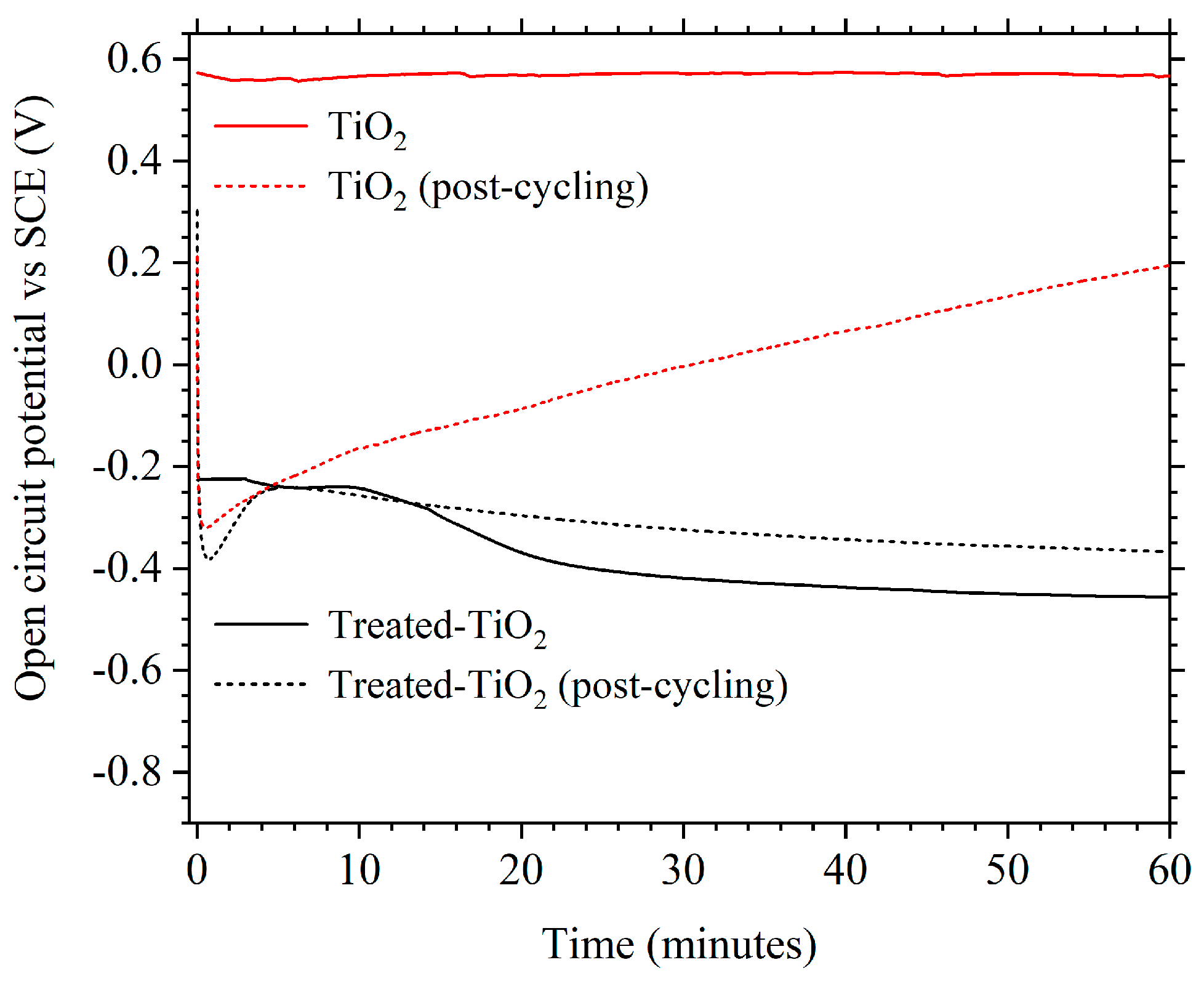
3.2. Electrochemical Performance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Feng, Z.; Laul, D.; Zhu, W.; Provencher, M.; Trudeau, M.L.; Guerfi, A.; Zaghib, K. Ultra-low cost and highly stable hydrated FePO4 anodes for aqueous sodium-ion battery. J. Power Sources 2018, 374, 211–216. [Google Scholar] [CrossRef]
- Li, W.; Dahn, J.R.; Wainwright, D.S. Rechargeable Lithium Batteries with Aqueous Electrolytes. Science 1994, 264, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hong, J.; Park, K.-Y.; Kim, H.; Kim, S.-W.; Kang, K. Aqueous Rechargeable Li and Na Ion Batteries. Chem. Rev. 2014, 114, 11788–11827. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-P.; Wang, P.-F.; Wang, T.-S.; Yin, Y.-X.; Guo, Y.-G.; Wang, C.-R. Prussian blue nanocubes as cathode materials for aqueous Na-Zn hybrid batteries. J. Power Sources 2017, 355, 18–22. [Google Scholar] [CrossRef]
- Holland, A.; Mckerracher, R.D.; Cruden, A.; Wills, R.G.A. An aluminium battery operating with an aqueous electrolyte. J. Appl. Electrochem. 2018, 48, 243–250. [Google Scholar] [CrossRef]
- Kazazi, M.; Abdollahi, P.; Mirzaei-Moghadam, M. High surface area TiO2 nanospheres as a high-rate anode material for aqueous aluminium-ion batteries. Solid State Ion. 2017, 300, 32–37. [Google Scholar] [CrossRef]
- He, Y.J.; Peng, J.F.; Chu, W.; Li, Y.Z.; Tong, D.G. Black mesoporous anatase TiO2 nanoleaves: A high capacity and high rate anode for aqueous Al-ion batteries. J. Mater. Chem. A 2014, 2, 1721–1731. [Google Scholar] [CrossRef]
- Lahan, H.; Boruah, R.; Hazarika, A.; Das, S.K. Anatase TiO2 as an Anode Material for Rechargeable Aqueous Aluminum-Ion Batteries: Remarkable Graphene Induced Aluminum Ion Storage Phenomenon. J. Phys. Chem. C 2017, 121, 26241–26249. [Google Scholar] [CrossRef]
- Liu, Y.; Sang, S.; Wu, Q.; Lu, Z.; Liu, K.; Liu, H. The electrochemical behavior of Cl− assisted Al3+ insertion into titanium dioxide nanotube arrays in aqueous solution for aluminum ion batteries. Electrochim. Acta 2014, 143, 340–346. [Google Scholar] [CrossRef]
- Liu, S.; Hu, J.J.; Yan, N.F.; Pan, G.L.; Li, G.R.; Gao, X.P. Aluminum storage behavior of anatase TiO2 nanotube arrays in aqueous solution for aluminum ion batteries. Energy Environ. Sci. 2012, 5, 9743–9746. [Google Scholar] [CrossRef]
- Holland, A.W.; McKerracher, R.; Cruden, A.; Wills, R.G.A. TiO2 nanopowder as a high rate, long cycle life electrode in aqueous aluminium electrolyte. Mater. Today Energy 2018, 10, 208–213. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, T.; Chen, C.; Ling, M.; Lin, Z.; Zhang, S.; Pan, F.; Liang, C. High-performance aqueous symmetric sodium-ion battery using NASICON-structured Na2VTi(PO4)3. Nano Res. 2018, 11, 490–498. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, S.; Dong, Z.; Shang, Y. 1D nanostructured sodium vanadium oxide as a novel anode material for aqueous sodium ion batteries. Nano Energy 2014, 4, 49–55. [Google Scholar] [CrossRef]
- Whitacre, J.F.; Wiley, T.; Shanbhag, S.; Wenzhuo, Y.; Mohamed, A.; Chun, S.E.; Weber, E.; Blackwood, D.; Lynch-Bell, E.; Gulakowski, J.; et al. An aqueous electrolyte, sodium ion functional, large format energy storage device for stationary applications. J. Power Sources 2012, 213, 255–264. [Google Scholar] [CrossRef]
- Zhang, B.H.; Liu, Y.; Chang, Z.; Yang, Y.Q.; Wen, Z.B.; Wu, Y.P.; Holze, R. Nanowire Na0.35MnO2 from a hydrothermal method as a cathode material for aqueous asymmetric supercapacitors. J. Power Sources 2014, 253, 98–103. [Google Scholar] [CrossRef]
- Gang, P.; Ping, N.; Changzhou, Y.; Laifa, S.; Xiaogang, Z.; Jiajia, Z.; Bing, D. Enhanced Performance of Aqueous Sodium-Ion Batteries Using Electrodes Based on the NaTi2(PO4)3/MWNTs–Na0.44MnO2 System. Energy Technol. 2014, 2, 705–712. [Google Scholar]
- Liang, C.; Leyuan, Z.; Xufeng, Z.; Zhaoping, L. Aqueous Batteries Based on Mixed Monovalence Metal Ions: A New Battery Family. ChemSusChem 2014, 7, 2295–2302. [Google Scholar]
- Yao, H.-R.; You, Y.; Yin, Y.-X.; Wan, L.-J.; Guo, Y.-G. Rechargeable dual-metal-ion batteries for advanced energy storage. Phys. Chem. Chem. Phys. 2016, 18, 9326–9333. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Song, B.; Zhang, J.; Ma, H. A rechargeable Na-Zn hybrid aqueous battery fabricated with nickel hexacyanoferrate and nanostructured zinc. J. Power Sources 2016, 321, 257–263. [Google Scholar] [CrossRef]
- Takai, O. Solution plasma processing (SPP). Pure Appl. Chem. 2008, 80, 2003. [Google Scholar] [CrossRef]
- Pitchaimuthu, S.; Honda, K.; Suzuki, S.; Naito, A.; Suzuki, N.; Katsumata, K.-I.; Nakata, K.; Ishida, N.; Kitamura, N.; Idemoto, Y.; et al. Solution Plasma Process-Derived Defect-Induced Heterophase Anatase/Brookite TiO2 Nanocrystals for Enhanced Gaseous Photocatalytic Performance. ACS Omega 2018, 3, 898–905. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Huang, F. Black titanium dioxide (TiO2) nanomaterials. Chem. Soc. Rev. 2015, 44, 1861–1885. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, G.; Zhai, T.; Yu, M.; Gan, J.; Tong, Y.; Li, Y. Hydrogenated TiO2 Nanotube Arrays for Supercapacitors. Nano Lett. 2012, 12, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Li, S.; Gray, E.; Liu, H.; Gu, Q.-F.; Sun, C.; Lai, C.; Zhao, H.; Zhang, S. Hydrogenation Synthesis of Blue TiO2 for High-Performance Lithium-Ion Batteries. J. Phys. Chem. C 2014, 118, 8824–8830. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Joo, J.H.; Samuelis, D.; Maier, J. Oxygen-Deficient TiO2−δ Nanoparticles via Hydrogen Reduction for High Rate Capability Lithium Batteries. Chem. Mater. 2012, 24, 543–551. [Google Scholar] [CrossRef]
- He, H.; Yang, K.; Wang, N.; Luo, F.; Chen, H. Hydrogenated TiO2 film for enhancing photovoltaic properties of solar cells and self-sensitized effect. J. Appl. Phys. 2013, 114, 213505. [Google Scholar] [CrossRef]
- Song, J.; Zheng, M.; Yuan, X.; Li, Q.; Wang, F.; Ma, L.; You, Y.; Liu, S.; Liu, P.; Jiang, D.; et al. Electrochemically induced Ti3+ self-doping of TiO2 nanotube arrays for improved photoelectrochemical water splitting. J. Mater. Sci. 2017, 52, 6976–6986. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y. Electrochemically Self-Doped TiO2 Nanotube Arrays for Supercapacitors. J. Phys. Chem. C 2014, 118, 5626–5636. [Google Scholar] [CrossRef]
- Nah, Y.-C.; Paramasivam, I.; Schmuki, P. Doped TiO2 and TiO2 Nanotubes: Synthesis and Applications. ChemPhysChem 2010, 11, 2698–2713. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, C.; Wang, N.; Li, Y.; Ding, H. Stable Ti3+ Self-Doped Anatase-Rutile Mixed TiO2 with Enhanced Visible Light Utilization and Durability. J. Phys. Chem. C 2016, 120, 6116–6124. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, B.; Pan, R.; Yao, J.; Qiu, J.; Luo, L.; Liu, Y. Safe and facile hydrogenation of commercial Degussa P25 at room temperature with enhanced photocatalytic activity. RSC Adv. 2014, 4, 1128–1132. [Google Scholar] [CrossRef]
- Leshuk, T.; Parviz, R.; Everett, P.; Krishnakumar, H.; Varin, R.A.; Gu, F. Photocatalytic Activity of Hydrogenated TiO2. ACS Appl. Mater. Interfaces 2013, 5, 1892–1895. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Wen, Z.; Cui, S.; Hou, Y.; Guo, X.; Chen, J. Controllable Synthesis and Tunable Photocatalytic Properties of Ti3+-doped TiO2. Sci. Rep. 2015, 5, 10714. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ni, Y.; Lu, C.; Xu, Z. Hydrogenation temperature related inner structures and visible-light-driven photocatalysis of N–F co-doped TiO2 nanosheets. Appl. Surf. Sci. 2014, 290, 125–130. [Google Scholar] [CrossRef]
- Gelderman, K.; Lee, L.; Donne, S.W. Flat-Band Potential of a Semiconductor: Using the Mott–Schottky Equation. J. Chem. Educ. 2007, 84, 685. [Google Scholar] [CrossRef]
- Sellers, M.C.K.; Seebauer, E.G. Measurement method for carrier concentration in TiO2 via the Mott–Schottky approach. Thin Solid Films 2011, 519, 2103–2110. [Google Scholar] [CrossRef]
- Yadav, P.; Pandey, K.; Bhatt, P.; Tripathi, B.; Pandey, M.K.; Kumar, M. Probing the electrochemical properties of TiO2/graphene composite by cyclic voltammetry and impedance spectroscopy. Mater. Sci. Eng. B 2016, 206, 22–29. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holland, A.; McKerracher, R.; Cruden, A.; Wills, R. Electrochemically Treated TiO2 for Enhanced Performance in Aqueous Al-Ion Batteries. Materials 2018, 11, 2090. https://doi.org/10.3390/ma11112090
Holland A, McKerracher R, Cruden A, Wills R. Electrochemically Treated TiO2 for Enhanced Performance in Aqueous Al-Ion Batteries. Materials. 2018; 11(11):2090. https://doi.org/10.3390/ma11112090
Chicago/Turabian StyleHolland, Alexander, Rachel McKerracher, Andrew Cruden, and Richard Wills. 2018. "Electrochemically Treated TiO2 for Enhanced Performance in Aqueous Al-Ion Batteries" Materials 11, no. 11: 2090. https://doi.org/10.3390/ma11112090
APA StyleHolland, A., McKerracher, R., Cruden, A., & Wills, R. (2018). Electrochemically Treated TiO2 for Enhanced Performance in Aqueous Al-Ion Batteries. Materials, 11(11), 2090. https://doi.org/10.3390/ma11112090