
Bioactive Glass-Ceramic Scaffolds from Novel ‘Inorganic Gel Casting’ and Sinter-Crystallization

 ,
,  and
and 
Abstract
:1. Introduction
2. Experimental Procedure
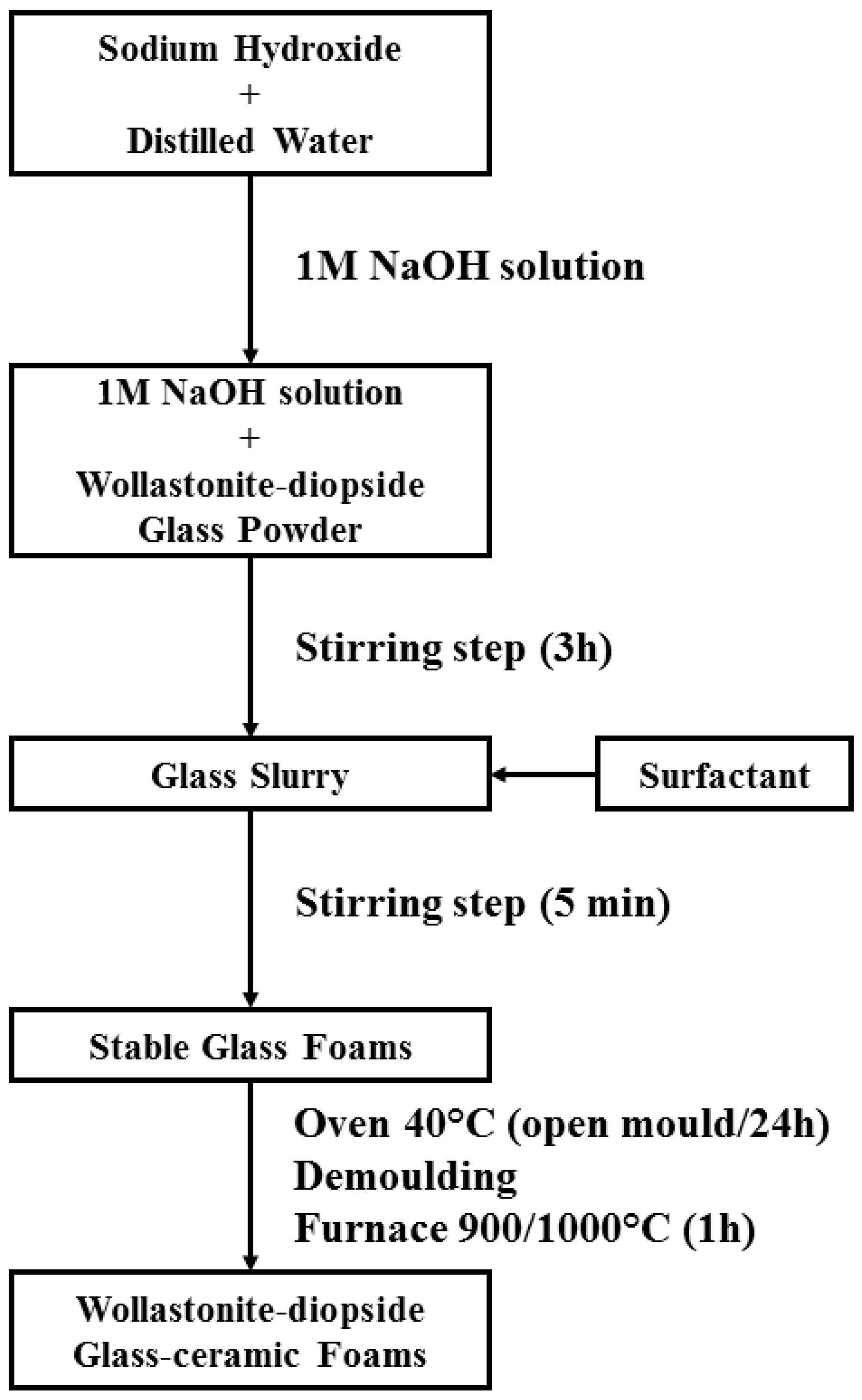
2.1. Starting Glass
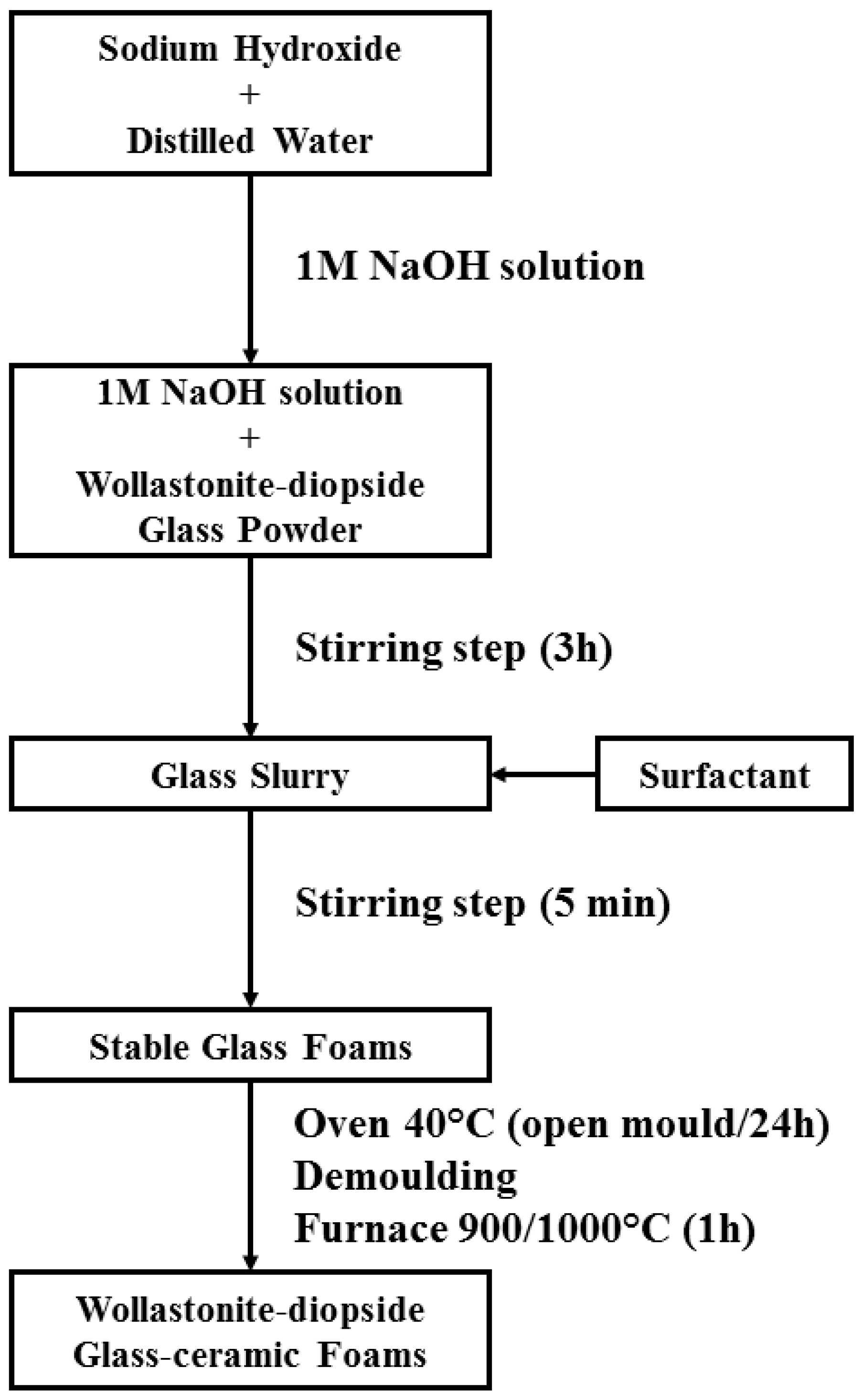
2.2. Preparation and Microstructural Characterization of Foams
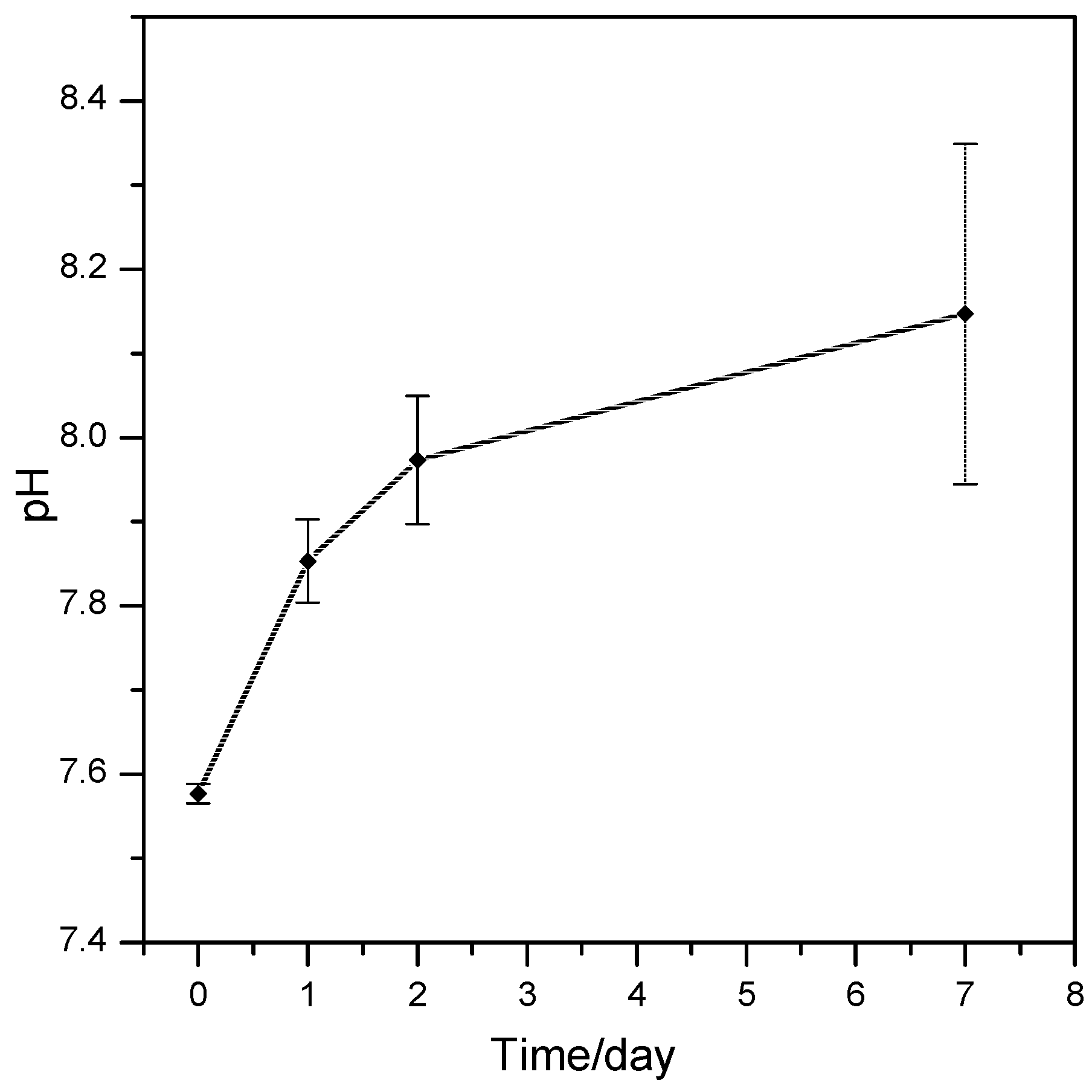
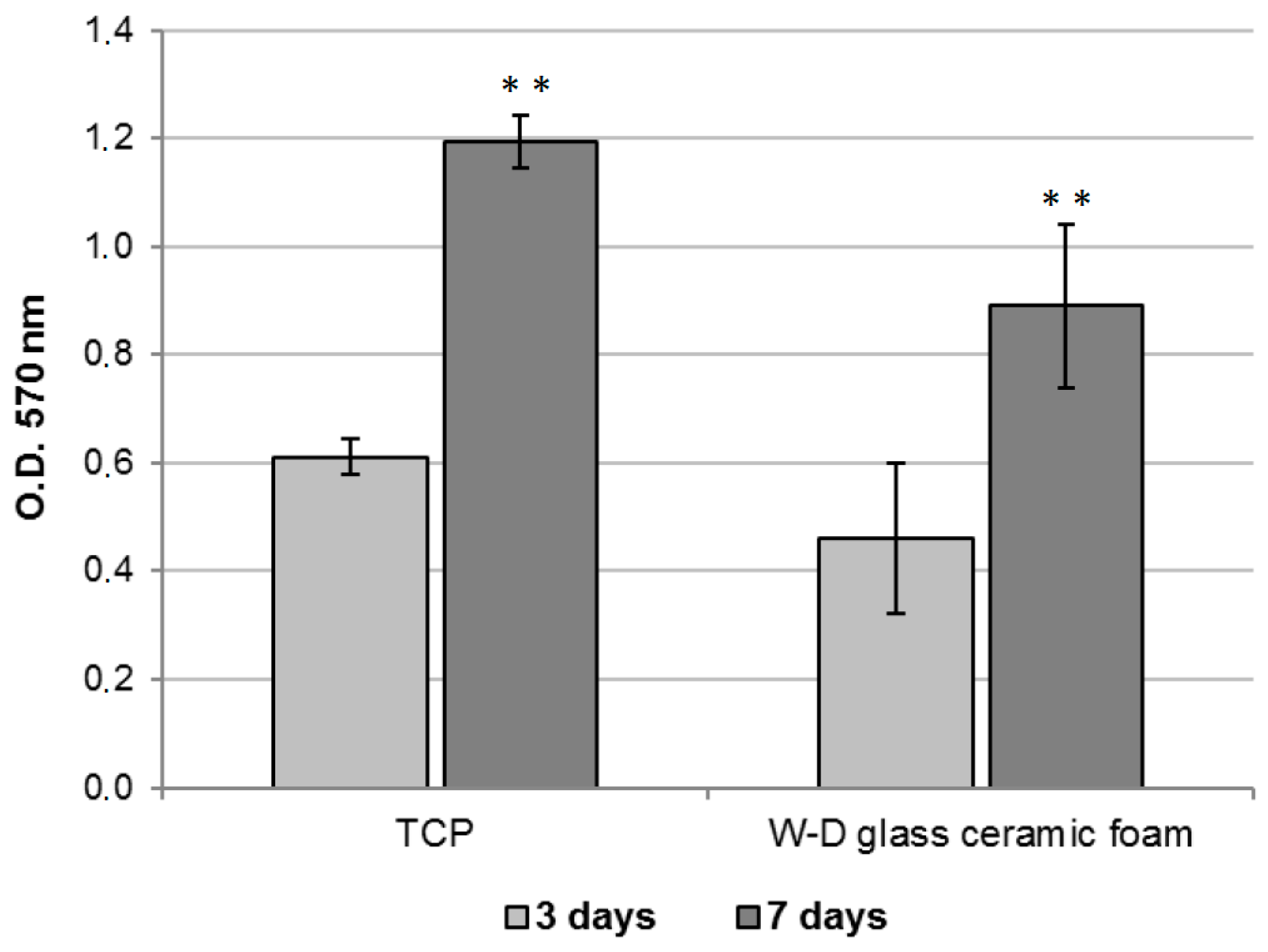
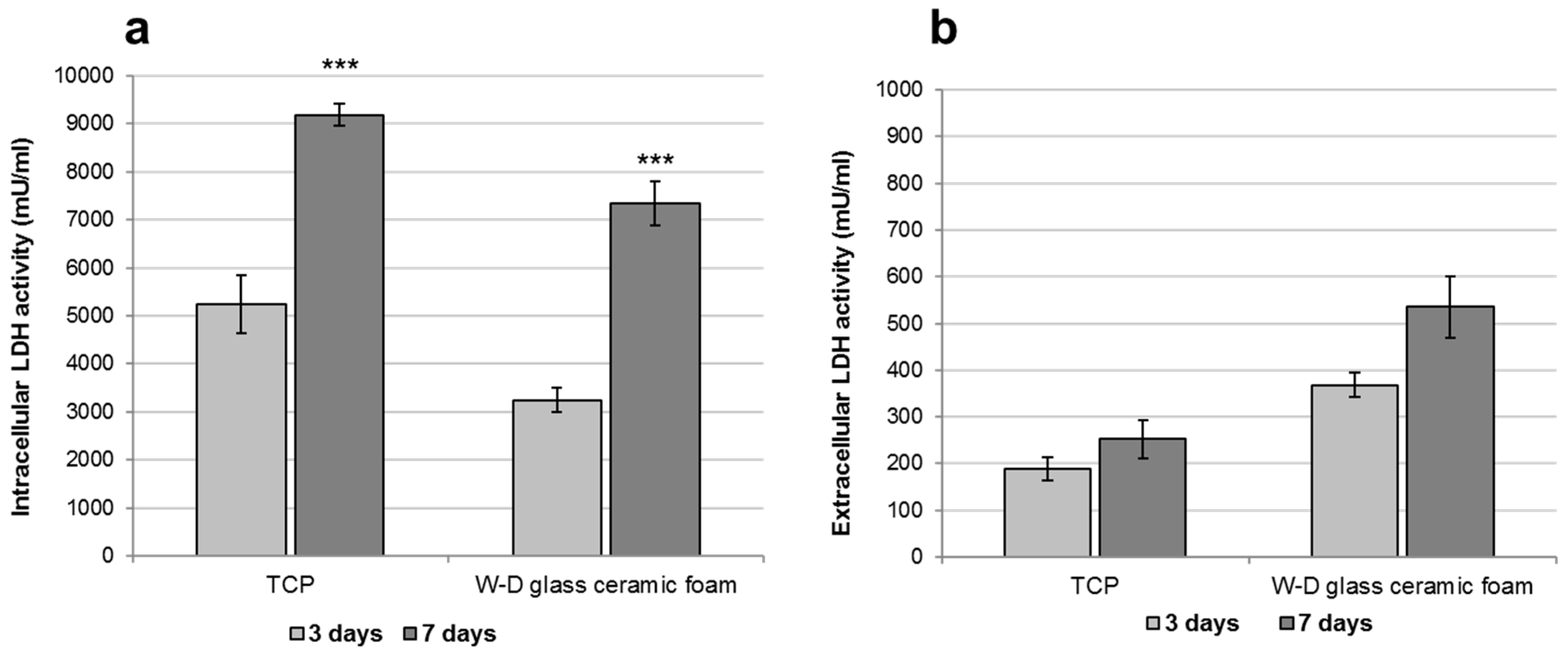
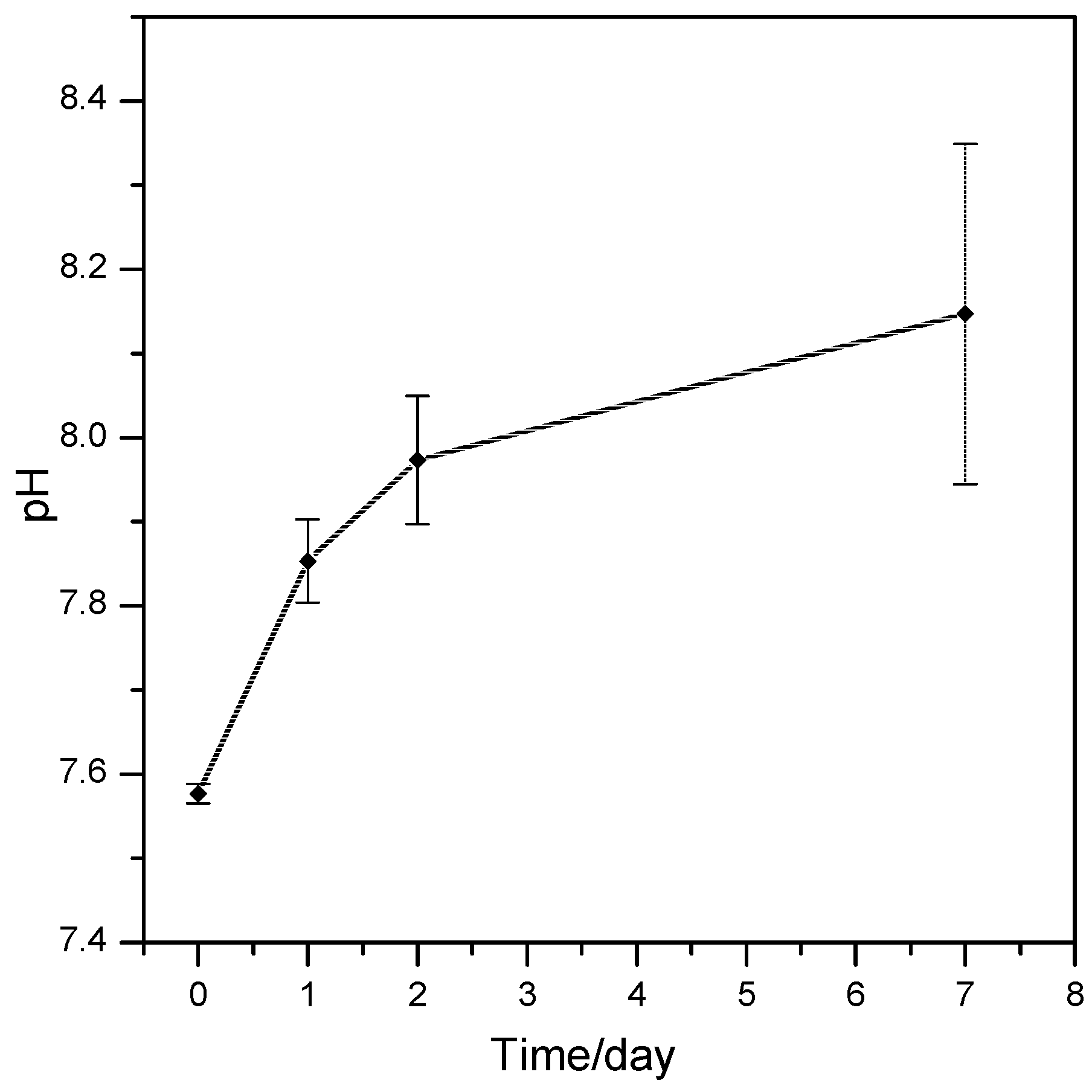
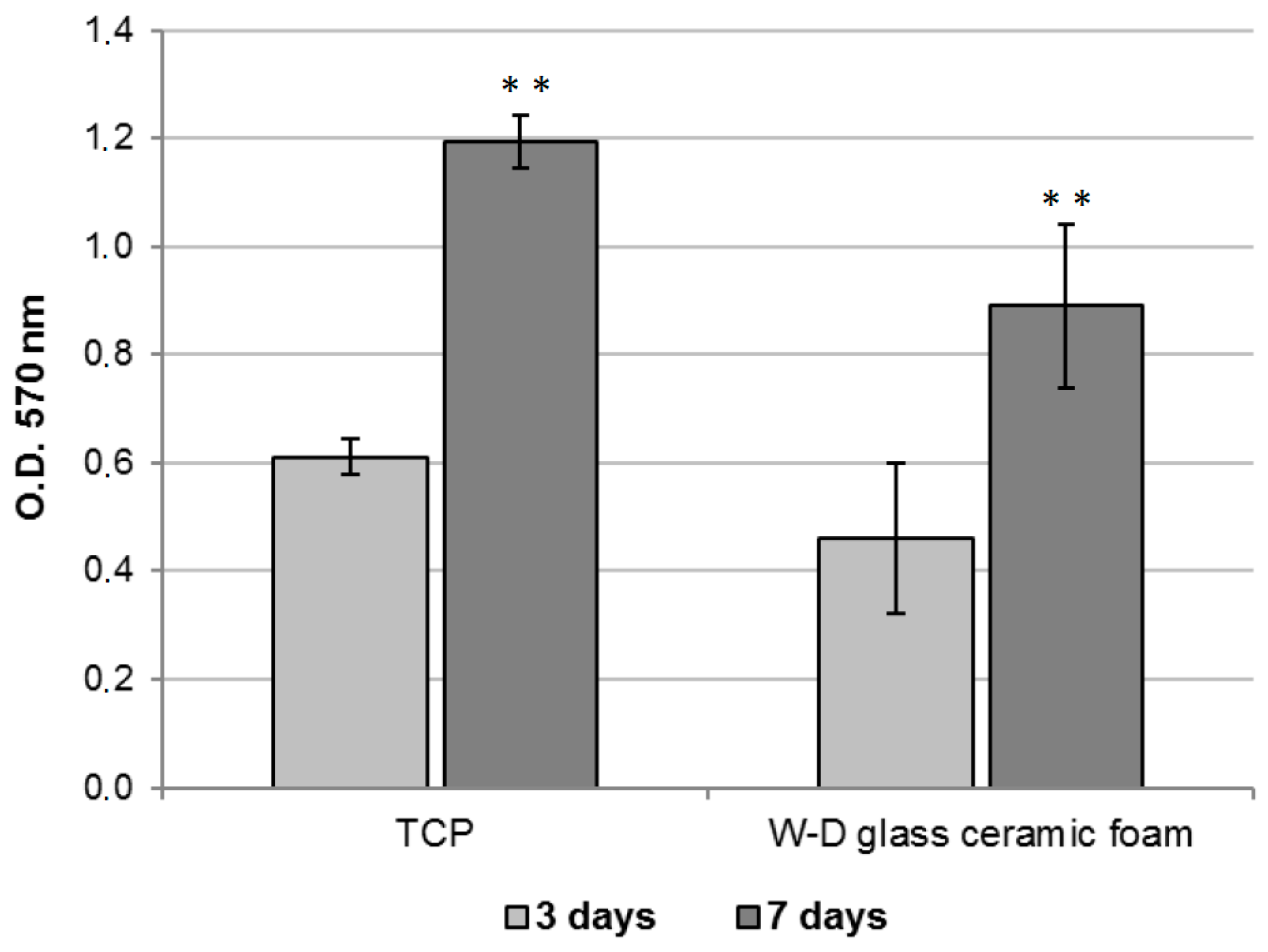
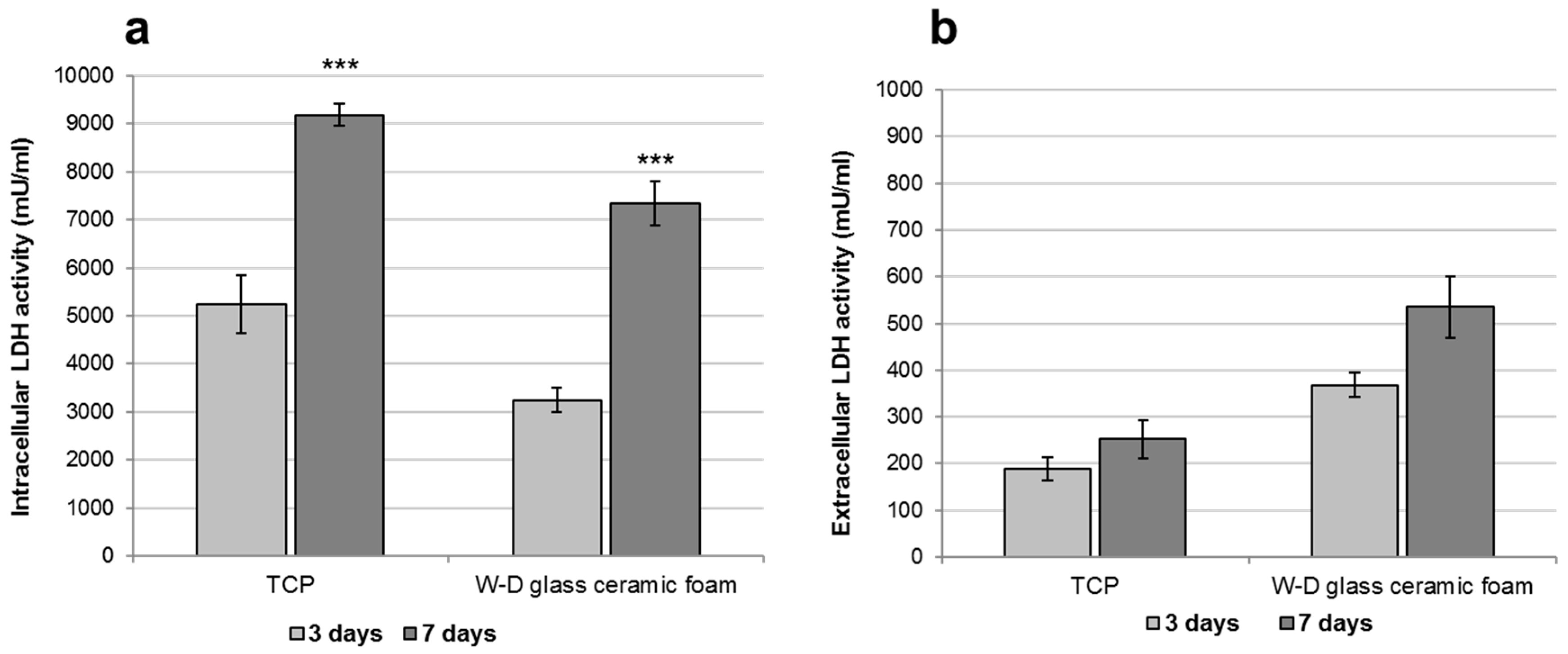
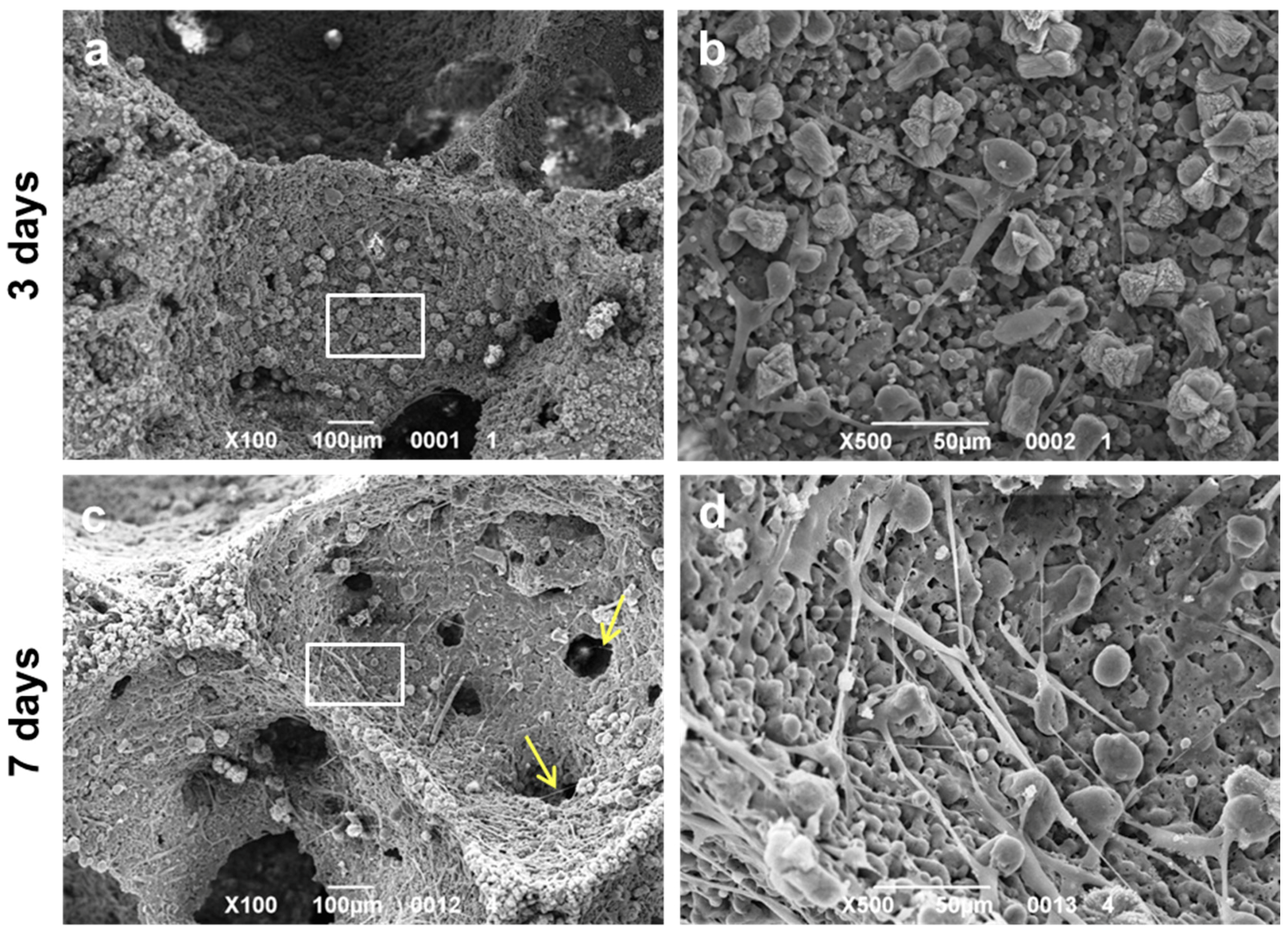
2.3. Assessment of the In Vitro Bioactivity and Cell Culture Test
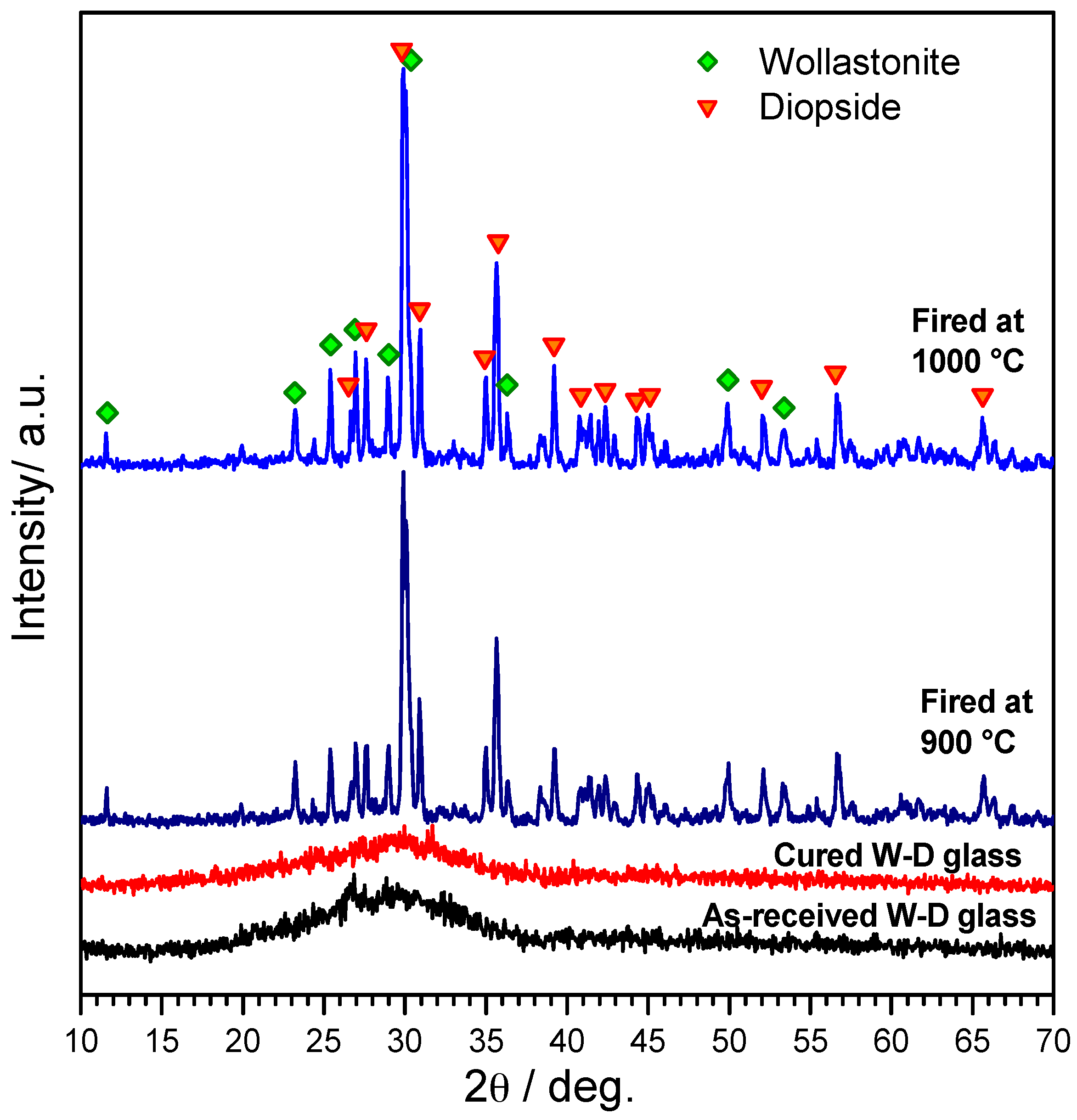
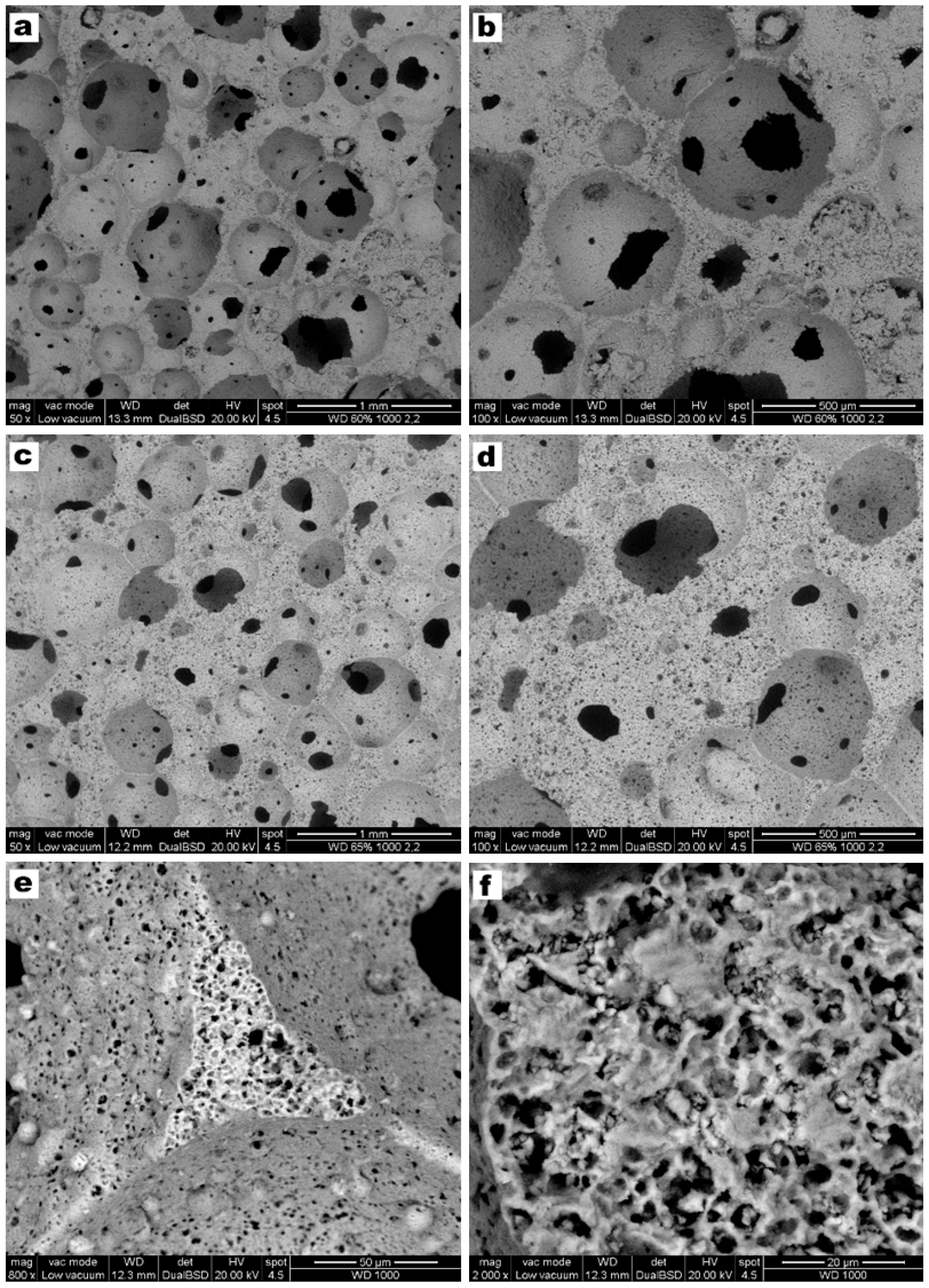
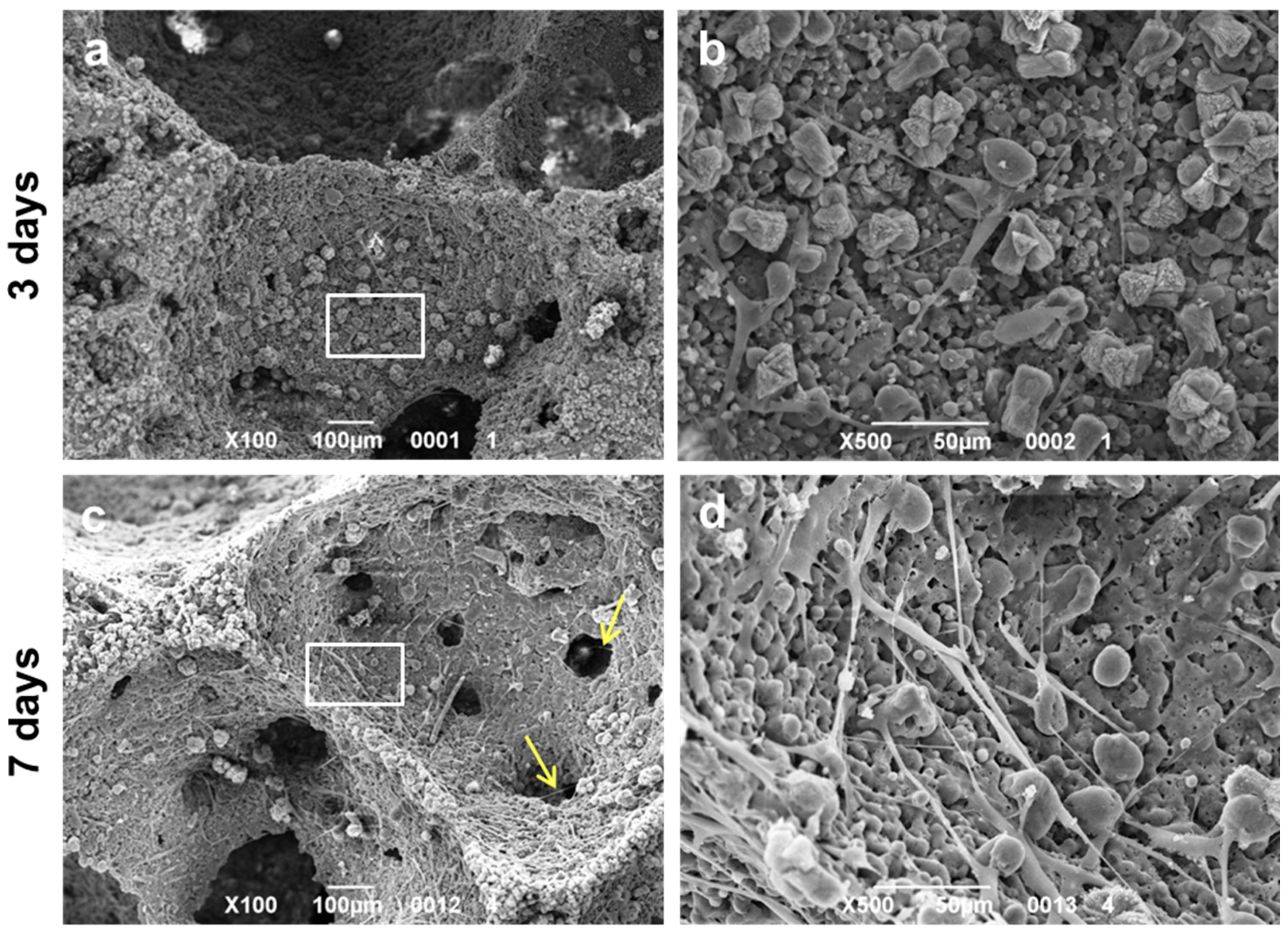
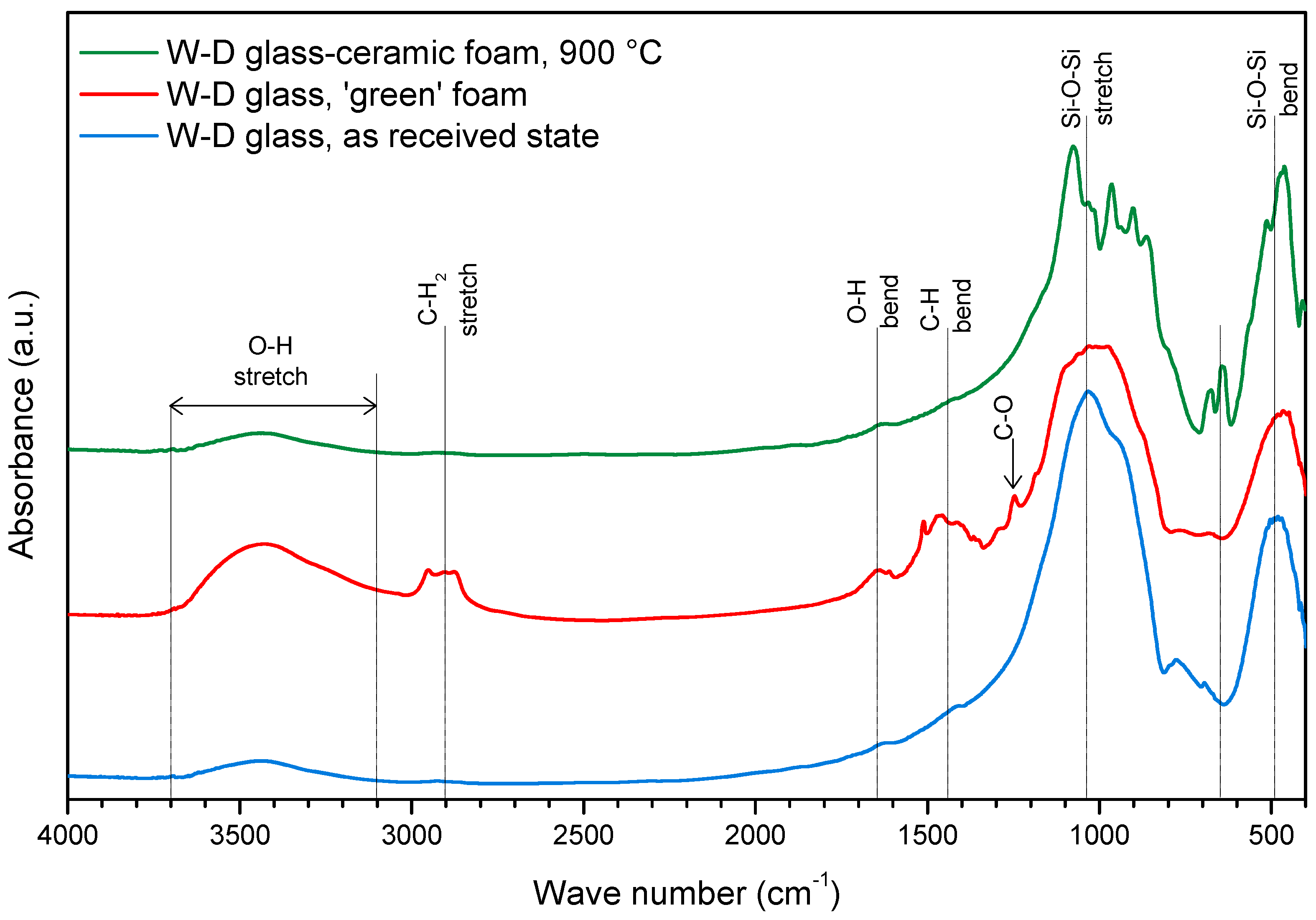
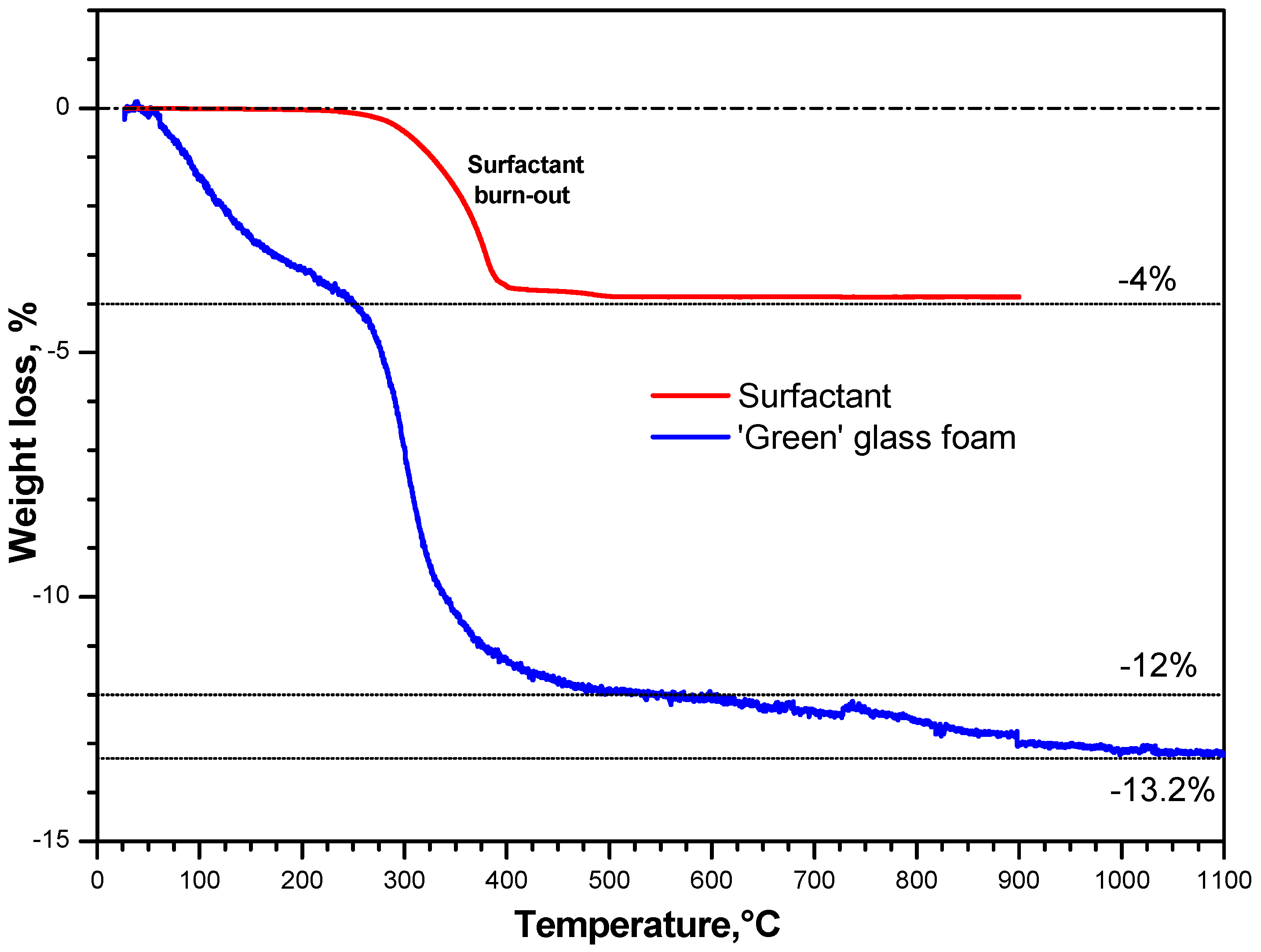
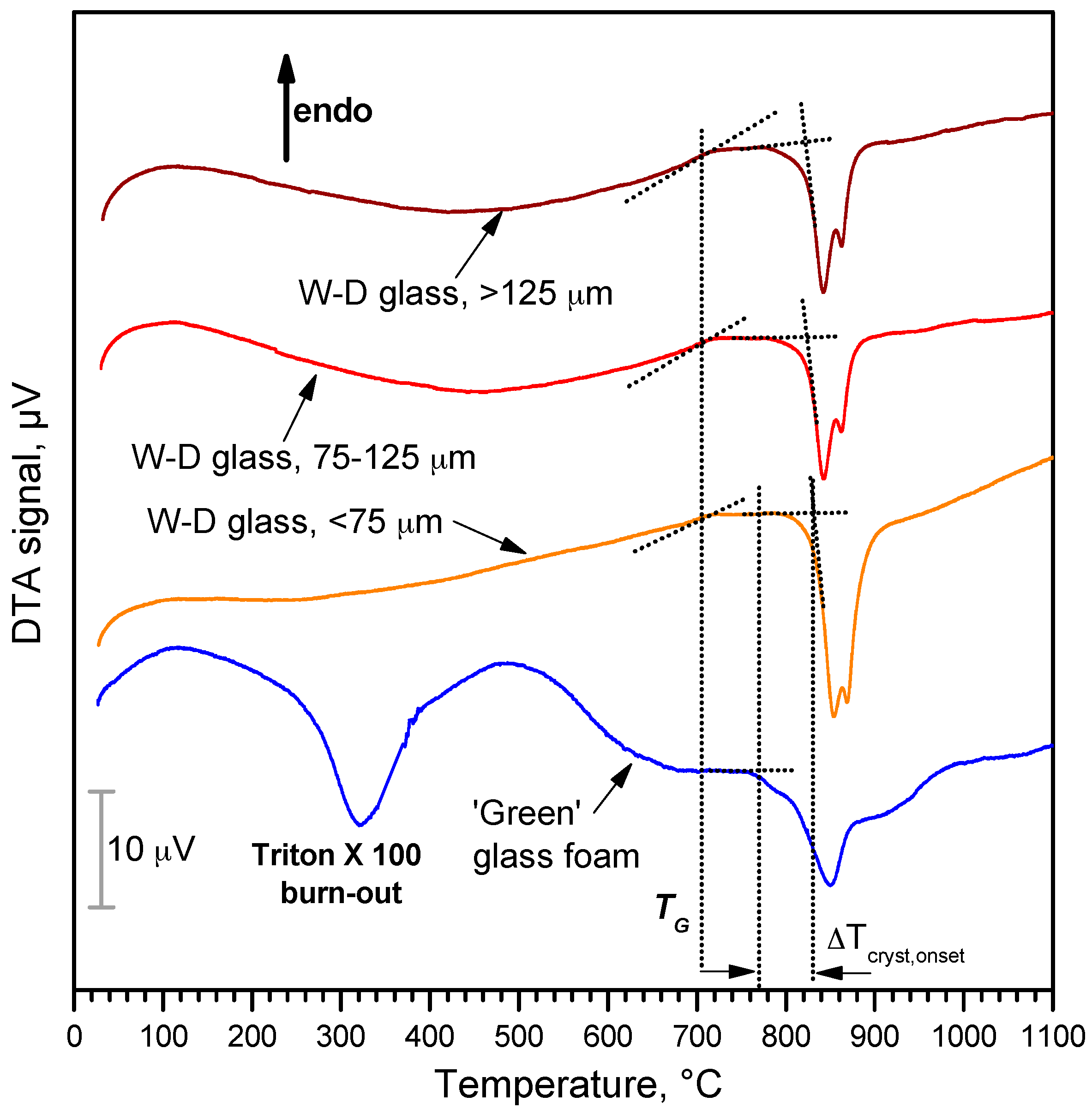
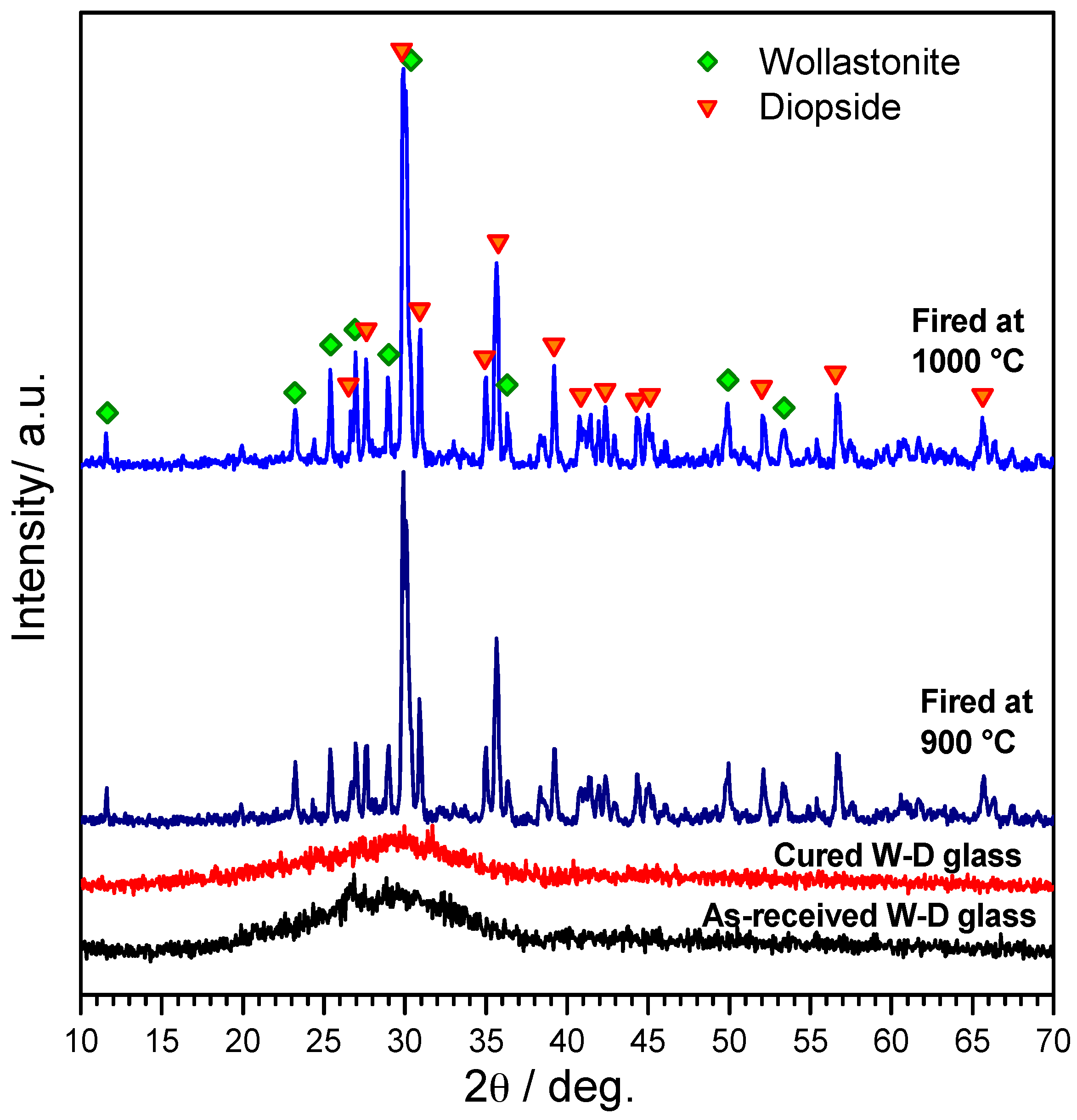
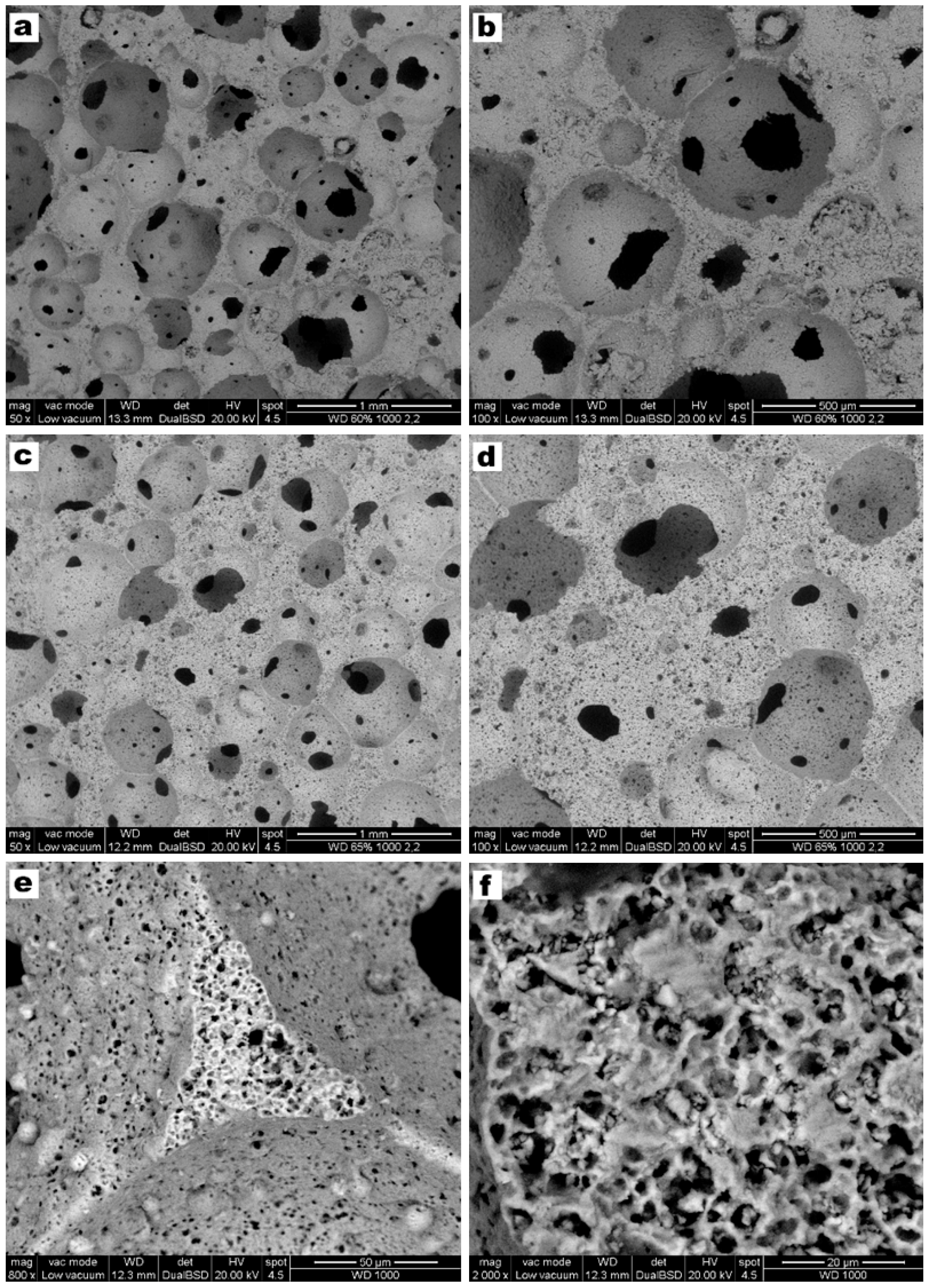
3. Results and Discussion
4. Conclusions
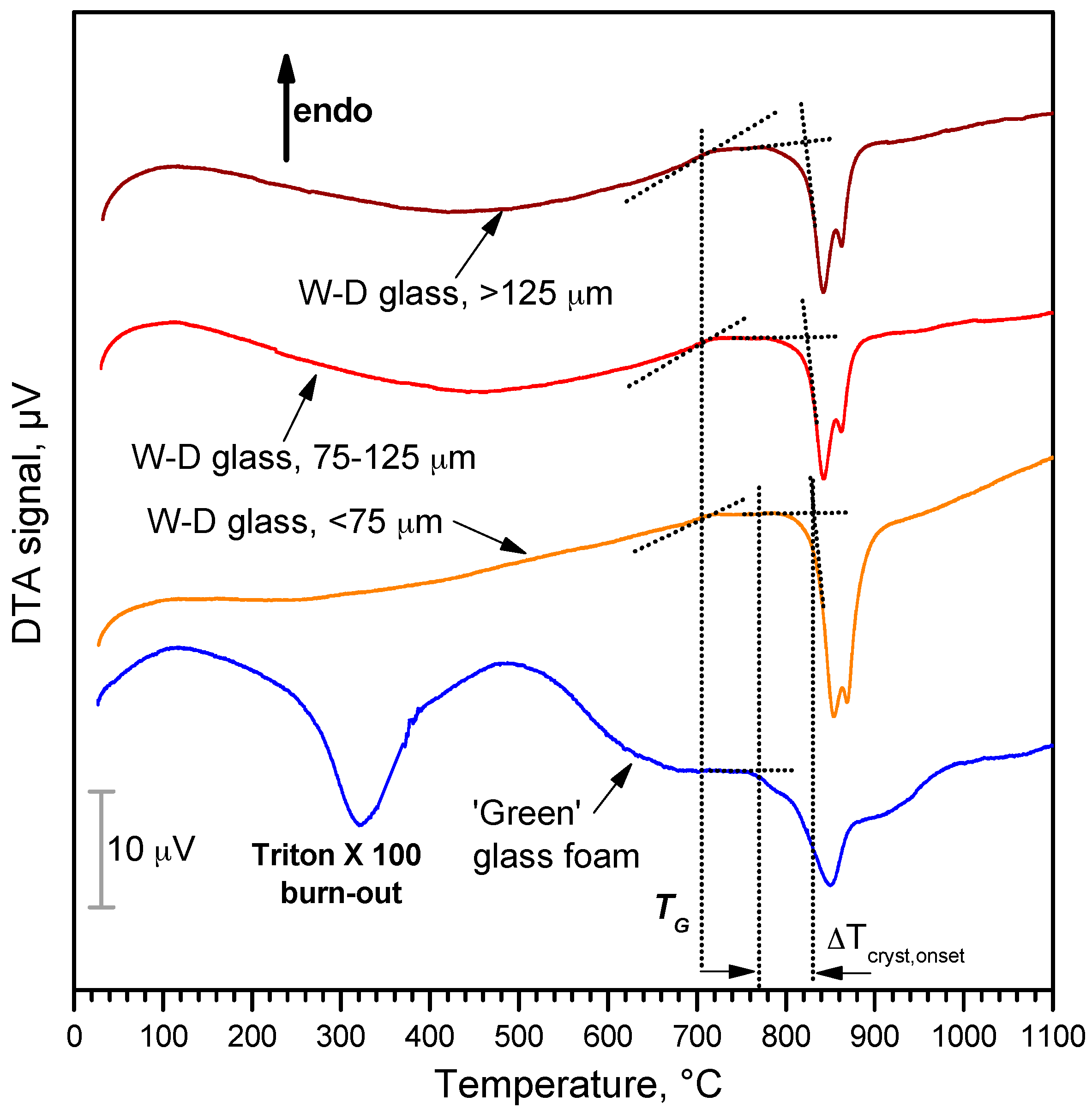
- Highly porous wollastonite-diopside glass-ceramics can be easily obtained by low temperature ‘inorganic gel-casting’, followed by sintering with concurrent crystallization (sinter-crystallization); the crystallization limits the viscous flow, so that the microstructure in the green state is substantially maintained upon firing up to 1000 °C;
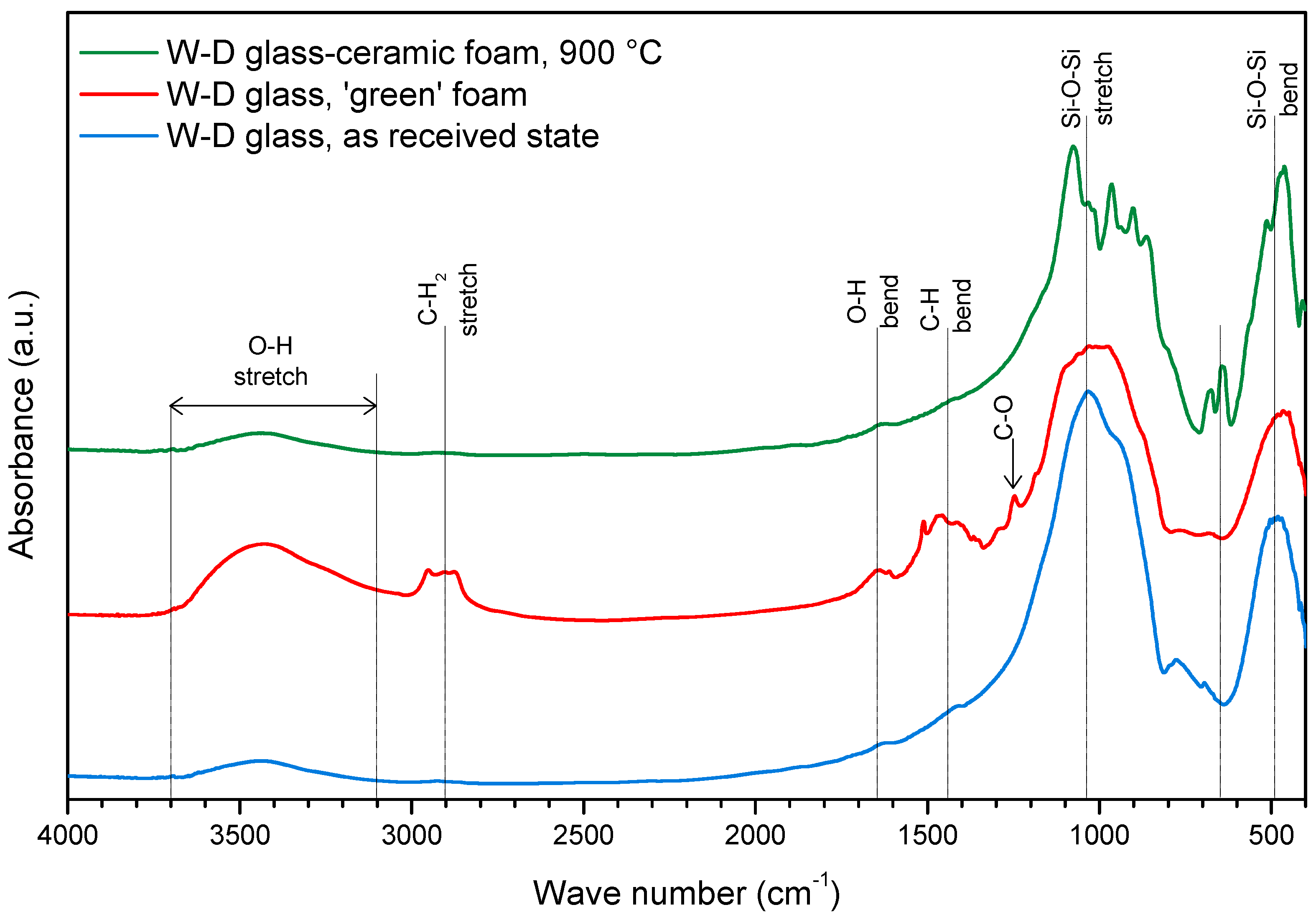
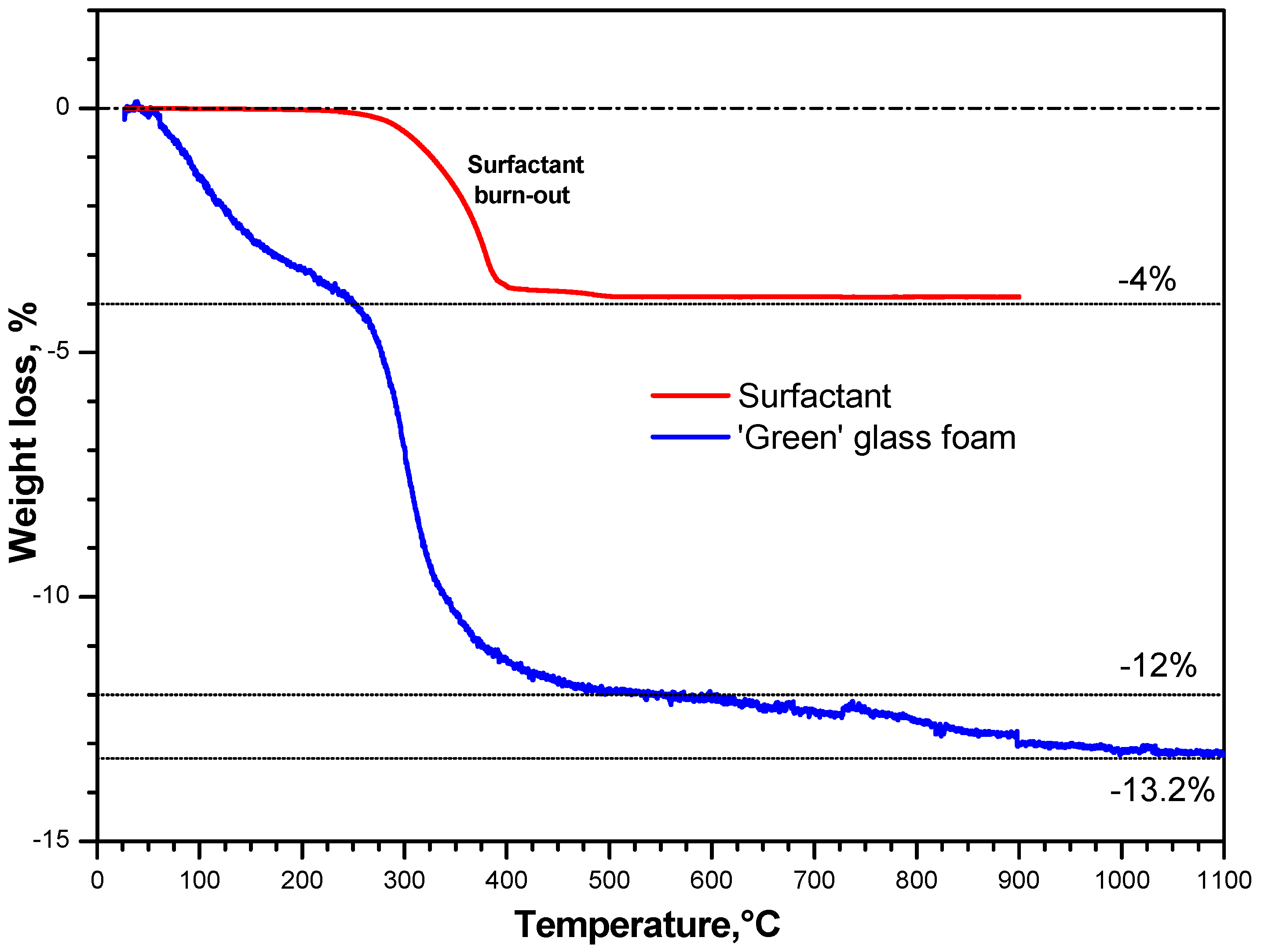
- The foaming relies on the progressive hardening of aqueous glass suspensions, after alkali activation; the gelification, owing to FTIR analysis, is consistent with the development of calcium silicate hydrates (C–S–H), later decomposed (with the firing treatment);
- The overall process (mechanical stirring of alkali activated suspensions—with the help of a surfactant, drying, firing with sinter-crystallization) has a great potential for the production of ‘hierarchically porous’ foams; the microstructure can be tuned operating on simple processing parameters such solid load, in suspensions, and firing conditions (e.g., heating rate);
- The developed glass-ceramics, according to MTT and LDH activity assays, with human fibroblasts, can be considered as biocompatible; forthcoming studies will focus on detailed studies of ionic releases and bioactivity.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Aza, P.N.; Guitian, F.; De Aza, S. Bioactivity of wollastonite ceramics: In vitro evaluation. Scr. Metall. Mater. 1994, 31, 1001–1005. [Google Scholar] [CrossRef]
- Lin, K.; Zhai, W.; Ni, S.; Chang, J.; Zeng, Y.; Qian, W. Study of mechanical property and in vitro biocompatibility of CaSiO3 ceramics. Ceram. Int. 2005, 31, 323–326. [Google Scholar] [CrossRef]
- Wu, C.; Chang, J. Degradation, bioactivity and cytocompatibility of diopside, akermanite and bredigite ceramics. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 83, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Nonami, T.; Tsutsumi, S. Study of diopside ceramics for biomaterials. J. Mater. Sci. Mater. Med. 1999, 10, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ramaswamy, Y.; Zreiqat, H. Porous diopside (CaMgSi2O6) scaffold: A promising bioactive material for bone tissue engineering. Acta Biomater. 2010, 6, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Sainz, M.A.; Pena, O.; Serena, S.; Caballero, A. Influence of design on bioactivity of novel CaSiO3-CaMg(SiO3)2 bioceramics: In vitro simulated body fluid test and thermodynamic simulation. Acta Biomater. 2010, 6, 2797–2807. [Google Scholar] [CrossRef] [PubMed]
- Tulyaganov, D.U.; Agathopoulos, S.; Ferreira, J.M.F. Sintering and crystallization of akermanite-based glass–ceramics. Mater. Lett. 2006, 60, 1488–1491. [Google Scholar]
- Agathopoulos, S.; Tulyaganov, D.U.; Ventura, J.M.G.; Kannan, S.; Karakassides, M.A.; Ferreira, J.M.F. Formation of hydroxyapatite onto glasses of the CaO-MgO-SiO2 system with B2O3, Na2O, CaF2 and P2O5 additives. Biomaterials 2006, 27, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Kansal, I.; Tulyaganov, D.U.; Goel, A.; Pascual, M.J.; Ferreira, J.M.F. Structural analysis and thermal behavior of diopside–fluorapatite–wollastonite-based glasses and glass–ceramics. Acta Biomater. 2010, 6, 4380–4388. [Google Scholar] [CrossRef] [PubMed]
- Tulyaganov, D.U.; Agathopoulos, S.; Valerio, P.; Balamurugan, A.; Saranti, A.; Karakassides, M.A.; Ferreira, J.M.F. Synthesis, bioactivity and preliminary biocompatibility studies of glasses in the system CaO-MgO-SiO2-Na2O-P2O5-CaF2. J. Mater. Sci. Mater. Med. 2011, 22, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, S.F.; Morrison, S.J.; Klawitter, J.J. Tissue reaction to three ceramics of porous and non-porous structures. J. Biomed. Mater. Res. 1972, 6, 347–374. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.J.; Ashby, M.F. Cellular Solids, Structure and Properties, 2nd ed.; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Vitale-Brovarone, C.; Baino, F.; Verné, E. High strength bioactive glass-ceramic scaffolds for bone regeneration. J. Mater. Sci. Mater. Med. 2009, 20, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, L.C.; Boccaccini, A.R. Bioactive glass and glass-ceramic scaffolds for bone tissue engineering. Materials 2010, 3, 3867–3910. [Google Scholar] [CrossRef]
- Baino, F.; Vitale-Brovarone, C. Bioactive glass and glass–ceramic foam scaffolds for bone tissue restoration. In Biomedical Foams for Tissue Engineering Applications; Netti, P., Ed.; Woodhead Publishing: Cambridge, UK, 2014; pp. 213–248. [Google Scholar]
- Baino, F.; Ferraris, M.; Bretcanu, O.; Verné, E.; Vitale-Brovarone, C. Optimization of composition, structure and mechanical strength of bioactive 3-D glass-ceramic scaffolds for bone substitution. J. Appl. Biomater. 2013, 27, 872–890. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, P.; Jones, J.R.; Hench, L.L. In vitro dissolution of melt derived 45S5 and sol-gel derived 58S bioactive glasses. J. Biomed. Mater. Res. 2002, 61, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Hench, L.L. Effect of surfactant concentration and composition on the structure and properties of sol-gel-derived bioactive glass scaffolds for tissue engineering. J. Mater. Sci. 2003, 38, 1–8. [Google Scholar] [CrossRef]
- Jones, J.R.; Hench, L.L. The effect of processing variables on the properties of bioactive glass foams. J. Biomed. Mater. Res. B 2004, 68, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Gowsihan Poologasundarampillai, G.; Lee, P.D.; Lam, C.; Kourkouta, A.M.; Jones, J.R. Compressive strength of bioactive sol–gel glass foam scaffolds. Int. J. Appl. Glass Sci. 2016, 7, 229–237. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Hill, R.G.; Yue, S.; Nightingale, D.; Lee, P.D.; Jones, J.R. Melt-derived bioactive glass scaffolds produced by a gel-cast foaming technique. Acta Biomater. 2011, 7, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Novajra, G.; Perdika, P.; Pisano, R.; Miola, M.; Bari, A.; Jones, J.R.; Detsch, R.; Boccaccini, A.R.; Vitale-Brovarone, C. Structure optimisation and biological evaluation of bone scaffolds prepared by co-sintering of silicate and phosphate glasses. Adv. Appl. Ceram. 2015, 114, S48–S55. [Google Scholar] [CrossRef]
- Provis, J.L. Geopolymers and other alkali activated materials: Why, how, and what? Mater. Struct. 2014, 47, 11–25. [Google Scholar] [CrossRef]
- Garbev, K.; Black, L.; Beuchle, G.; Stemmermann, P. Inorganic polymers in cement based materials. Wasser-und Geotechnol. 2002, 1, 19–30. [Google Scholar]
- Garcia-Lodeiro, I.; Fernández-Jimenez, A.; Pena, P.; Palomo, A. Alkaline activation of synthetic aluminosilicate glass. Ceram. Int. 2014, 40, 5547–5558. [Google Scholar] [CrossRef]
- Garcia-Lodeiro, I.; Aparicio-Rebollo, E.; Fernández-Jimenez, A.; Palomo, A. Effect of calcium on the alkaline activation of aluminosilicate glass. Ceram. Int. 2016, 42, 7697–7707. [Google Scholar] [CrossRef]
- Ruiz-Santaquiteria, C.; Fernández-Jiménez, A.; Palomo, A. Alternative prime materials for developing new cements: Alkaline activation of alkali aluminosilicate glasses. Ceram. Int. 2016, 42, 9333–9340. [Google Scholar] [CrossRef]
- Puertas, F.; Torres-Carrasco, M. Use of glass waste as an activator in the preparation of alkali-activated slag. Mechanical strength and paste characterisation. Cem. Concr. Res. 2014, 57, 95–104. [Google Scholar] [CrossRef]
- Redden, R.; Neithalath, N. Microstructure, strength, and moisture stability of alkali activated glass powder-based binders. Cem. Concr. Compos. 2014, 45, 46–56. [Google Scholar] [CrossRef]
- Torres-Carrasco, M.; Puertas, F. Waste glass in the geopolymer preparation. Mechanical and microstructural characterisation. J. Clean. Prod. 2015, 90, 397–408. [Google Scholar] [CrossRef]
- Cyr, M.; Idir, R.; Poinot, T. Properties of inorganic polymer (geopolymer) mortars made of glass cullet. J. Mater. Sci. 2012, 47, 2782–2797. [Google Scholar] [CrossRef]
- Rincón, A.; Giacomello, G.; Pasetto, M.; Bernardo, E. Novel ‘inorganic gel casting’ process for the manufacturing of glass foams. J. Eur. Ceram. Soc. 2017, 37, 2227–2234. [Google Scholar] [CrossRef]
- Strozi Cilla, M.; Colombo, P.; Morelli, M.R. Geopolymer foams by gelcasting. Ceram. Int. 2014, 40, 5723–5730. [Google Scholar] [CrossRef]
- Bernardo, E. Micro- and macro-cellular sintered glass-ceramics from wastes. J. Eur. Ceram. Soc. 2007, 27, 2415–2422. [Google Scholar] [CrossRef]
- Wang, X.; Ruan, J.; Chen, Q. Effects of surfactants on the microstructure of porous ceramic scaffolds fabricated by foaming for bone tissue engineering. Mater. Res. Bull. 2009, 44, 1275–1279. [Google Scholar] [CrossRef]
- Fiocco, L.; Elsayed, H.; Bernardo, E.; Daguano, J.K.M.F.; Soares, V.O. Silicone resins mixed with active oxide fillers and Ca-Mg silicate glass as alternative/integrative precursors for wollastonite-diopside glass-ceramic foams. J. Non-Cryst. Solids 2015, 416, 44–49. [Google Scholar] [CrossRef]
- Fiocco, L.; Elsayed, H.; Bernardo, E.; Ferroni, L.; Gardin, C.; Zavan, B. Bioactive wollastonite-diopside foams from preceramic polymers and reactive oxide fillers. Materials 2015, 8, 2480–2494. [Google Scholar] [CrossRef]
- Shah, A.T.; Batool, M.; Chaudhry, A.A.; Iqbal, F.; Javaid, A.; Zahid, S.; Ilyas, K.; Bin Qasim, S.; Khan, A.F.; Khan, A.S.; et al. Effect of calcium hydroxide on mechanical strength and biological properties of bioactive glass. J. Mech. Behav. Biomed. Mater. 2016, 61, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Chakradhar, R.S.; Nagabhushana, B.M.; Chandrappa, G.T.; Ramesh, K.P.; Rao, J.L. Solution combustion derived nanocrystalline macroporous wollastonite ceramics. Mater. Chem. Phys. 2006, 95, 169–175. [Google Scholar] [CrossRef]
- Monsivais-Gámez, E.; Ruiz, F.; Martínez, J.R. Four-membered rings family in the Si–O extended rocking IR band from quantum chemistry calculations. J. Sol-Gel Sci. Technol. 2007, 43, 65–72. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, G. Dehydration kinetics of Portland cement paste at high temperature. J. Therm. Anal. Calor. 2012, 110, 153–158. [Google Scholar] [CrossRef]
- Karamanov, A.; Pelino, M. Induced crystallization porosity and properties of sintered diopside and wollastonite glass-ceramics. J. Eur. Ceram. Soc. 2008, 28, 555–562. [Google Scholar] [CrossRef]
- Watanabe, T.; Hashimoto, H.; Hayashi, M.; Nagata, K. Effect of alkali oxides on crystallization in CaO-SiO2-CaF2 glasses. ISIJ Int. 2008, 48, 925–933. [Google Scholar] [CrossRef]
- Hemmings, R.; Berry, E. On the glass in coal fly ashes: Recent advances. In MRS Proceedings; Cambridge University Press: Cambridge, UK, 1987. [Google Scholar]
- Ni, S.; Chang, J.; Chou, L. A novel bioactive porous CaSiO3 scaffold for bone tissue engineerin’. J. Biomed. Mater. Res. A 2006, 76, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, H.; Sepantafar, M.; Ostadrahimi, A. The role of bioinorganics in improving the mechanical properties of silicate ceramics as bone regenerative materials. J. Ceram. Sci. Technol. 2015, 6, 1–8. [Google Scholar]
- Wu, C.; Chang, J. A review of bioactive silicate ceramics. Biomed. Mater. 2013, 8, 032001. [Google Scholar] [CrossRef] [PubMed]
- Kansal, I.; Goel, A.; Tulyaganov, D.U.; Pascual, M.J.; Lee, H.-Y.; Kim, H.-W.; Ferreira, J.M.F. Diopside (CaO·MgO·2SiO2)–fluorapatite(9CaO·3P2O5·CaF2) glass-ceramics: Potential materials for bone tissue engineering. J. Mater. Chem. 2011, 21, 16247–16256. [Google Scholar] [CrossRef]
- Jones, J.R.; Ehrenfried, L.M.; Hench, L.L. Optimising bioactive glass scaffolds for bone tissue engineering. Biomaterials 2006, 27, 964–973. [Google Scholar] [CrossRef] [PubMed]
- ImageJ—Image Processing and Analysis in Java. Available online: https://imagej.nih.gov/ij/ (accessed on 25 January 2016).
- Rahaman, M.N.; Day, D.E.; Bal, B.S.; Fu, Q.; Jung, S.B.; Bonewald, L.F.; Tomsia, A.P. Bioactive glass in tissue engineering. Acta Biomater. 2011, 7, 2355–2373. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solid Load | Heating Rate (°C/min), Up to 1000 °C | Bulk Density (g/cm3) | True Density (g/cm3) | Total Porosity (vol %) | Open Porosity (vol %) | Compressive Strength (MPa) |
|---|---|---|---|---|---|---|
| 60 wt % | 2 °C/min | 0.29 ± 0.02 | 2.94 ± 0.01 | 90.6 | 90.1 | 3.50 ± 0.51 |
| 5 °C/min | 0.42 ± 0.05 | 2.95 ± 0.02 | 85.6 | 83.8 | 2.17 ± 0.10 | |
| 65 wt % | 2 °C/min | 0.44 ± 0.03 | 2.97 ± 0.01 | 86.3 | 85.3 | 2.90 ± 0.50 |
| 5 °C/min | 0.53 ± 0.04 | 2.95 ± 0.01 | 81.9 | 81.1 | 5.30 ± 0.74 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsayed, H.; Rincón Romero, A.; Ferroni, L.; Gardin, C.; Zavan, B.; Bernardo, E. Bioactive Glass-Ceramic Scaffolds from Novel ‘Inorganic Gel Casting’ and Sinter-Crystallization. Materials 2017, 10, 171. https://doi.org/10.3390/ma10020171
Elsayed H, Rincón Romero A, Ferroni L, Gardin C, Zavan B, Bernardo E. Bioactive Glass-Ceramic Scaffolds from Novel ‘Inorganic Gel Casting’ and Sinter-Crystallization. Materials. 2017; 10(2):171. https://doi.org/10.3390/ma10020171
Chicago/Turabian StyleElsayed, Hamada, Acacio Rincón Romero, Letizia Ferroni, Chiara Gardin, Barbara Zavan, and Enrico Bernardo. 2017. "Bioactive Glass-Ceramic Scaffolds from Novel ‘Inorganic Gel Casting’ and Sinter-Crystallization" Materials 10, no. 2: 171. https://doi.org/10.3390/ma10020171
APA StyleElsayed, H., Rincón Romero, A., Ferroni, L., Gardin, C., Zavan, B., & Bernardo, E. (2017). Bioactive Glass-Ceramic Scaffolds from Novel ‘Inorganic Gel Casting’ and Sinter-Crystallization. Materials, 10(2), 171. https://doi.org/10.3390/ma10020171