Tuning Patchy Bonds Induced by Critical Casimir Forces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
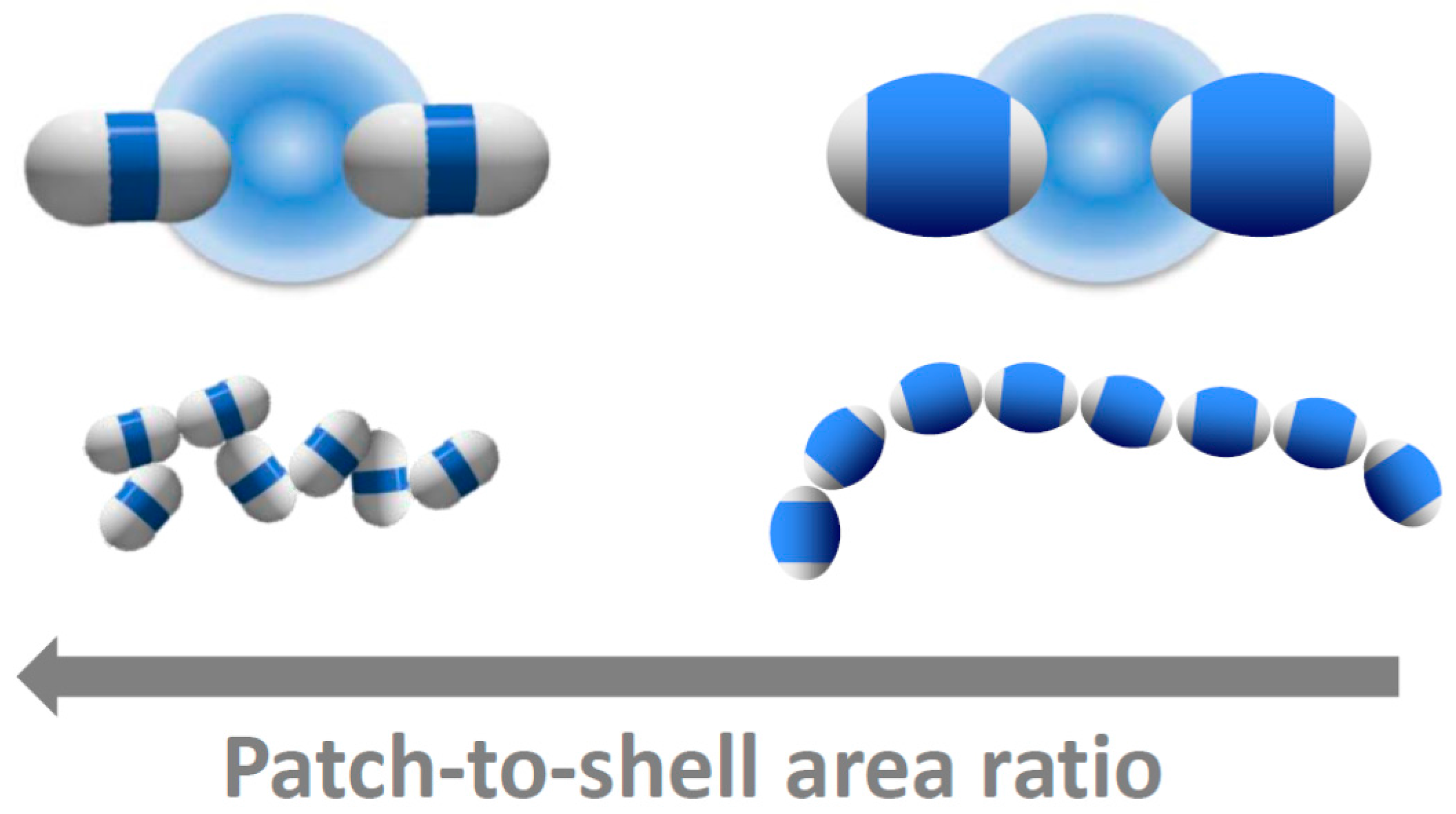{kind=link}
Abstract
1. Introduction
2. Experiment
3. Simulation
4. Results and Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vlasov, Y.A.; Bo, X.-Z.; Sturm, J.C.; Norris, D.J. On-chip natural assembly of silicon photonic bandgap crystals. Nature 2001, 414, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Hynninen, A.-P.; Thijssen, J.H.J.; Vermolen, E.C.M.; Dijkstra, M.; van Blaaderen, A. Self-assembly route for photonic crystals with a bandgap in the visible region. Nat. Mater. 2007, 6, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Kagan, C.R.; Murray, C.B. Charge transport in strongly coupled quantum dot solids. Nat. Nanotechnol. 2015, 10, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Lahann, J. Physical approaches to biomaterial design. Nat. Mater. 2009, 8, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lekkerkerker, H.N.W.; Tuinier, R. Colloids and the Depletion Interaction; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Leunissen, M.E.; Christova, C.G.; Hynninen, A.-P.; Royall, C.P.; Campbell, A.I.; Imhof, A.; Dijkstra, M.; van Roij, R.; van Blaaderen, A. Ionic colloidal crystals of oppositely charged particles. Nature 2005, 437, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.; Campbell, A.I. Three-dimensional binary superlattices of oppositely charged colloids. Phys. Rev. Lett. 2005, 95, 128302. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, E.V.; Talapin, D.V.; Kotov, N.A.; O’Brien, S.; Murray, C.B. Structural diversity in binary nanoparticle superlattices. Nature 2006, 439, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Zerrouki, D.; Baudry, J.; Pine, D.; Chaikin, P.; Bibette, J. Chiral colloidal clusters. Nature 2008, 455, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Sacanna, S.; Rossi, L.; Pine, D.J. Magnetic click colloidal assembly. J. Am. Chem. Soc. 2012, 134, 6112–6115. [Google Scholar] [CrossRef] [PubMed]
- Leunissen, M.E.; Dreyfus, R.; Cheong, F.C.; Grier, D.G.; Sha, R.; Seeman, N.C.; Chaikin, P.M. Switchable self-protected attractions in DNA-functionalized colloids. Nat. Mater. 2009, 8, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Nykypanchuk, D.; Maye, M.M.; van der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 2008, 451, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Breed, D.R.; Manoharan, V.N.; Feng, L.; Hollingsworth, A.D.; Weck, M.; Pine, D.J. Colloids with valence and specific directional bonding. Nature 2012, 491, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, V.N.; Elsesser, M.T.; Pine, D.J. Dense packing and symmetry in small clusters of microspheres. Science 2003, 301, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, S.C.; Solomon, M.J. Anisotropy of building blocks and their assembly into complex structures. Nat. Mater. 2007, 6, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Kraft, D.J.; Vlug, W.S.; van Kats, C.M.; van Blaaderen, A.; Imhof, A.; Kegel, W.K. Self-assembly of colloids with liquid protrusions. J. Am. Chem. Soc. 2009, 131, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Sacanna, S.; Korpics, M.; Rodriguez, K.; Kim, S.-H.; Pine, D.J.; Yi, G.-R. Shaping colloids for self-assembly. Nat. Commun. 2013, 4, 1688. [Google Scholar] [CrossRef] [PubMed]
- Kern, N.; Frenke, D. Fluid–fluid coexistence in colloidal systems with short-ranged strongly directional attraction. J. Chem. Phys. 2003, 118, 9882–9889. [Google Scholar] [CrossRef]
- Bianchi, E.; Largo, J.; Tartaglia, P.; Zaccarelli, E.; Sciortino, F. Phase diagram of patchy colloids: Towards empty liquids. Phys. Rev. Lett. 2006, 97, 168301. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Tartaglia, P.; Zaccarelli, E.; Sciortino, F. Theoretical and numerical study of the phase diagram of patchy colloids: Ordered and disordered patch arrangements. J. Chem. Phys. 2008, 128, 144504. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Cacciuto, A. Hierarchical self-assembly of asymmetric amphiphatic spherical colloidal particles. Phys. Rev. E 2009, 80, 021404. [Google Scholar] [CrossRef] [PubMed]
- Doppelbauer, G.; Bianchi, E.; Kahl, G. Self-assembly scenarios of patchy colloidal particles in two dimensions, J. Phys. Condens. Matter 2010, 22, 104105. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Lado, F.; Largo, J.; Pastore, G.; Sciortino, F. Effects of patch size and number within a simple model of patchy colloids. J. Chem. Phys. 2010, 132, 174110. [Google Scholar] [CrossRef] [PubMed]
- Munaò, G.; Preisler, Z.; Vissers, T.; Smallenburg, F.; Sciortino, F. Cluster formation in one-patch colloids: Low coverage results. Soft Matter 2013, 9, 2652–2661. [Google Scholar] [CrossRef]
- Smallenburg, F.; Sciortino, F. Liquids more stable than crystals in particles with limited valence and flexible bonds. Nat. Phys. 2013, 9, 554–558. [Google Scholar] [CrossRef]
- Fisher, M.E.; de Gennes, P.G. Phenomenes aux parois dans un melange binaire critique. C. R. Acad. Sci. Ser. B 1978, 287, 20–209. [Google Scholar]
- Beysens, D.; Estève, D. Adsorption phenomena at the surface of silica spheres in a binary liquid mixture. Phys. Rev. Lett. 1985, 54, 2123–2126. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Faber, S.; Hu, Z.; Wegdam, G.H.; Schall, P. Controlling colloidal phase transitions with critical Casimir forces. Nat. Commun. 2013, 4, 1584. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Newton, A.; Veen, S.J.; Kraft, D.J.; Bolhuis, P.G.; Schall, P. Switching colloidal superstructures by critical Casimir forces. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Hanke, A.; Schlesener, F.; Eisenriegler, E.; Dietrich, S. Critical casimir forces between spherical particles in fluids. Phys. Rev. Lett. 1998, 81, 1885. [Google Scholar] [CrossRef]
- Hertlein, C.; Helden, L.; Gambassi, A.; Dietrich, S.; Bechinger, C. Direct measurement of critical Casimir forces. Nature 2008, 451, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Elsesser, M.T.; Hollingsworth, A.D.; Edmond, K.V.; Pine, D.J. Large core-shell poly(methyl methacrylate) colloidal clusters: Synthesis, characterization, and tracking. Langmuir 2011, 27, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.D. Phase relationships in the pyridine series. Part II. The miscibility of some pyridine homologues with deuterium oxide. J. Chem. Soc. 1952, 4606–4608. [Google Scholar] [CrossRef]
- Stuij, S.G.; Labbé-Laurent, M.; Kodger, T.E.; Maciołek, A.; Schall, P. Critical Casimir interactions between colloids around the critical point of binary solvents. Soft Matter 2017, 13, 5233–5249. [Google Scholar] [CrossRef] [PubMed]
- Bonn, D.; Otwinowski, J.; Sacanna, S.; Guo, H.; Wegdam, G.H.; Schall, P. Direct observation of colloidal aggregation by critical Casimir forces. Phys. Rev. Lett. 2009, 103, 156101. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.T.; Vila Verde, A.; Bolhuis, P.G.; Schall, P. Temperature-sensitive colloidal phase behavior induced by critical Casimir forces. J. Chem Phys. 2013, 139, 094903. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C.; Nguyen, T.A.; Veen, S.J.; Kraft, D.J.; Schall, P.; Bolhuis, P.G. Modelling critical Casimir force induced self-assembly experiments on patchy colloidal dumbbells. Soft Matter 2017, 13, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Madhavan Unni, P.K. Critical behavior of the aqueous electrolytic system 3-methylpyridine + D2O + NaBr. J. Chem. Phys. 2006, 124, 054505. [Google Scholar] [CrossRef] [PubMed]
- Mohry, T.F.; Kondrat, S.; Maciolek, A.; Dietrich, S. Critical Casimir interactions around the consolute point of a binary solvent. Soft Matter 2014, 10, 5510–5522. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, K.W. Monte Carlo simulations of highly anisotropic two-dimensional hard dumbbell-shaped molecules: Nonperiodic phase between fluid and dense solid. Phys. Rev. B 1992, 46, 26. [Google Scholar] [CrossRef]
- Gerbode, S.J.; Lee, S.H.; Liddell, C.M.; Cohen, I. Restricted dislocation motion in crystals of colloidal dimer particles. Phys. Rev. Lett. 2008, 101, 058302. [Google Scholar] [CrossRef] [PubMed]
- Paladugu, S.; Callegari1, A.; Tuna, Y.; Barth, L.; Dietrich, S.; Gambassi, A.; Volpe, G. Nonadditivity of critical Casimir forces. Nat. Commun. 2016, 7, 11403. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.A.; Devailly, C.; Petrosyan, A.; Ciliberto, S. Energy transfer between colloids via critical interactions. Entropy 2017, 19, 77. [Google Scholar] [CrossRef]
- Palmiero, U.C.; Agostini, A.; Lattuada, E.; Gatti, S.; Singh, J.; Canova, C.T.; Buzzaccaro, S.; Moscatelli, D. Use of RAFT macro-surfmers for the synthesis of transparent aqueous colloids with tunable interactions. Soft Matter 2017, 13, 6439–6449. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.A.; Newton, A.; Kraft, D.J.; Bolhuis, P.G.; Schall, P. Tuning Patchy Bonds Induced by Critical Casimir Forces. Materials 2017, 10, 1265. https://doi.org/10.3390/ma10111265
Nguyen TA, Newton A, Kraft DJ, Bolhuis PG, Schall P. Tuning Patchy Bonds Induced by Critical Casimir Forces. Materials. 2017; 10(11):1265. https://doi.org/10.3390/ma10111265
Chicago/Turabian StyleNguyen, Truc A., Arthur Newton, Daniela J. Kraft, Peter G. Bolhuis, and Peter Schall. 2017. "Tuning Patchy Bonds Induced by Critical Casimir Forces" Materials 10, no. 11: 1265. https://doi.org/10.3390/ma10111265
APA StyleNguyen, T. A., Newton, A., Kraft, D. J., Bolhuis, P. G., & Schall, P. (2017). Tuning Patchy Bonds Induced by Critical Casimir Forces. Materials, 10(11), 1265. https://doi.org/10.3390/ma10111265