Catalytic Methane Decomposition for the Simultaneous Production of Hydrogen and Low-Reactivity Biocarbon for the Metallurgic Industry

 , ,
, , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Biocarbon Production
2.2. Methane Cracking Setups
2.3. Biocarbon Characterization
2.3.1. Proximate Analysis
2.3.2. Ultimate Analysis
2.3.3. Ash-Forming Element Analysis
2.3.4. Surface Area, Porosity and Density Analyses
2.3.5. Raman Analysis
2.3.6. SEM-EDS Analysis
2.3.7. Mechanical Property Analysis
2.3.8. CO2 Reactivity Procedure
3. Results and Discussion
3.1. Hydrogen Potential from Catalytic Methane Cracking
3.2. Characterization of the Treated Biocarbon
3.2.1. Proximate and Element Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proximate Analysis | Spruce Wood | Birch Wood | Wood Pellets | SEPs | Birch Bark |
|---|---|---|---|---|---|
| Biocarbon from pyrolysis | |||||
| Volatile matter content | 5.23 | 5.84 | 5.45 | 5.35 | 6.57 |
| Ash content | 0.82 | 1.29 | 0.80 | 1.08 | 3.98 |
| Fixed carbon content | 93.95 | 92.87 | 93.75 | 93.57 | 89.45 |
| Treated biocarbon, at 1100 °C, 90 min, and 90/10 (CH4/N2) | |||||
| Volatile matter content | 4.74 | 4.72 | 4.40 | 3.49 | 5.09 |
| Ash content | 0.79 | 1.09 | 0.77 | 1.03 | 2.08 |
| Fixed carbon content | 94.47 | 94.19 | 94.83 | 95.48 | 92.83 |
3.2.2. Ash-Forming Element Analysis
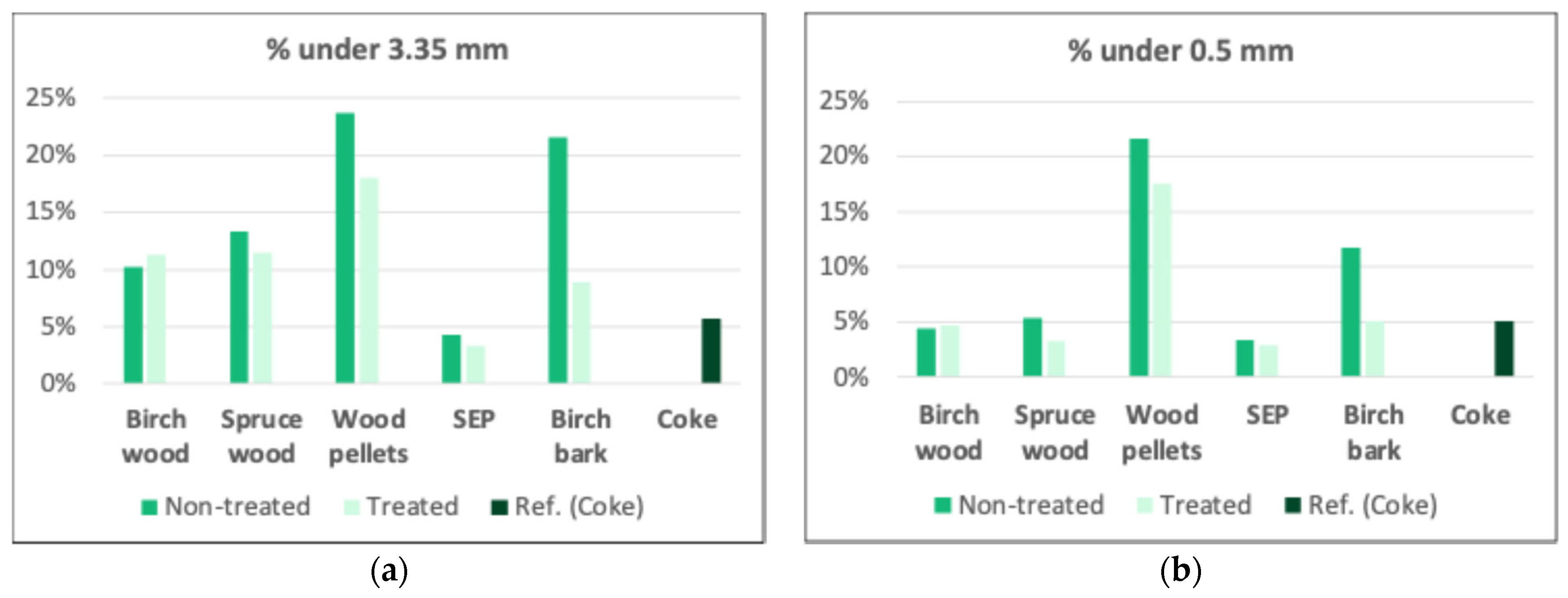
3.2.3. Surface and Density Analysis
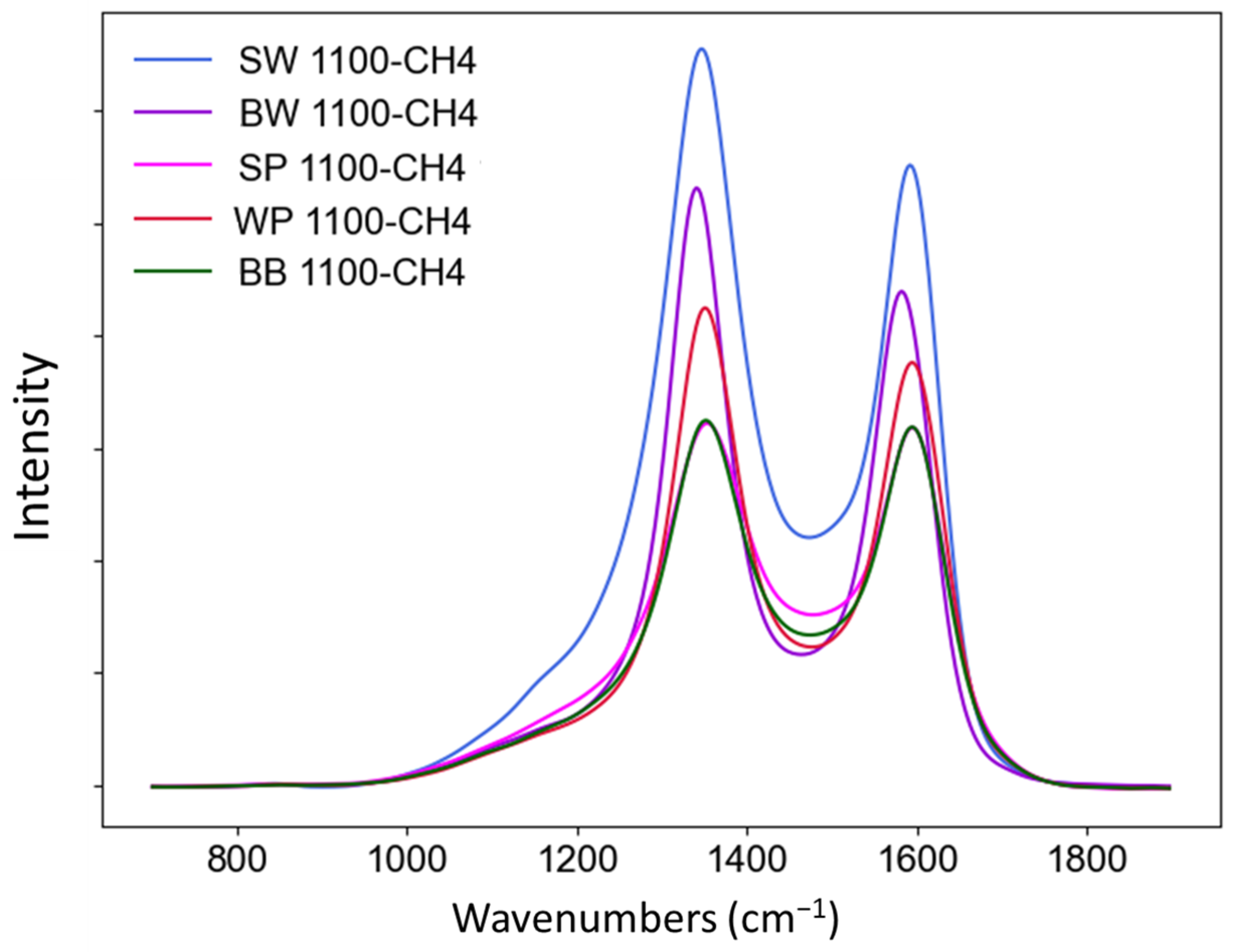
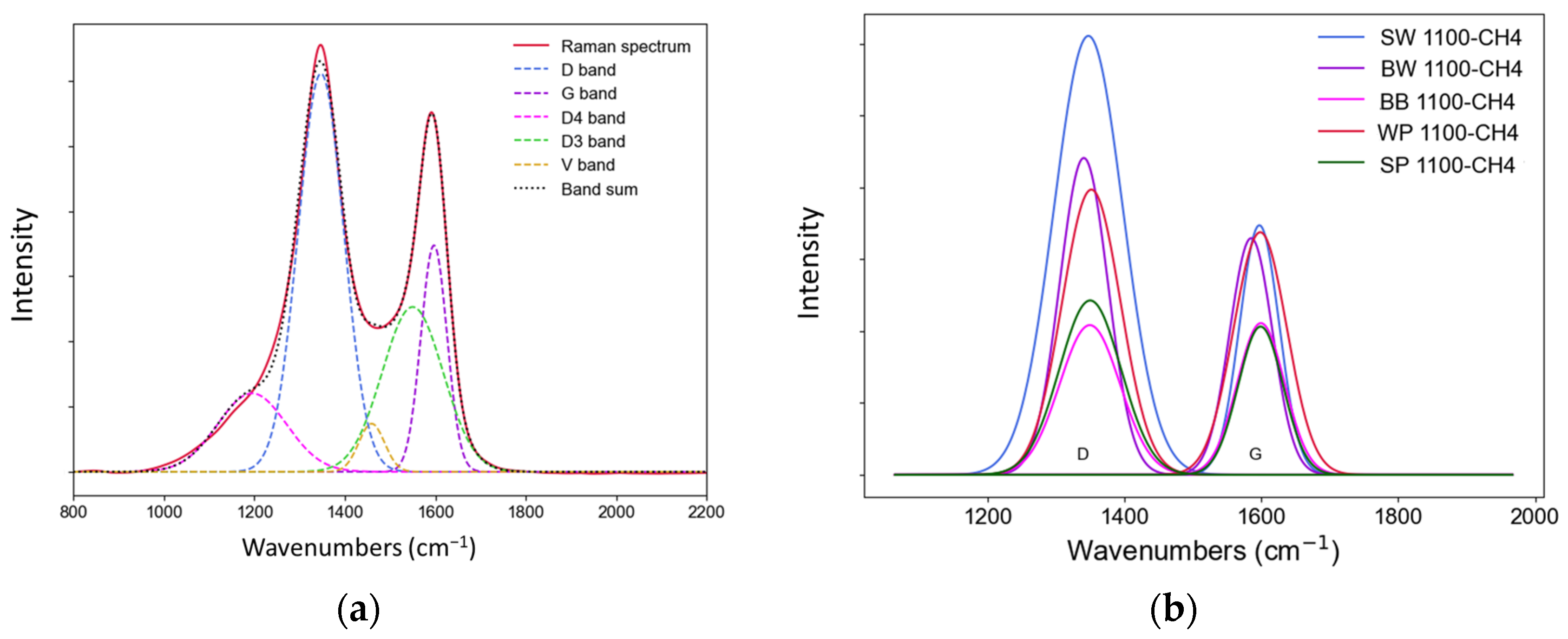
3.2.4. Raman Analysis
3.2.5. SEM-EDS Analysis
3.2.6. Mechanical Property Analysis
3.2.7. CO2 Reactivity
4. Conclusions
- The hydrogen production rate for all the tested materials was calculated as mol hydrogen produced per kg of biocarbon used. The tests were performed for a period of 90 min at a temperature of 1100 °C. Due to its high porosity, birch bark had the highest hydrogen production rate per catalyst mass at 105 mol/kg biocarbon. The lowest hydrogen production potential belonged to the SEP biocarbon.
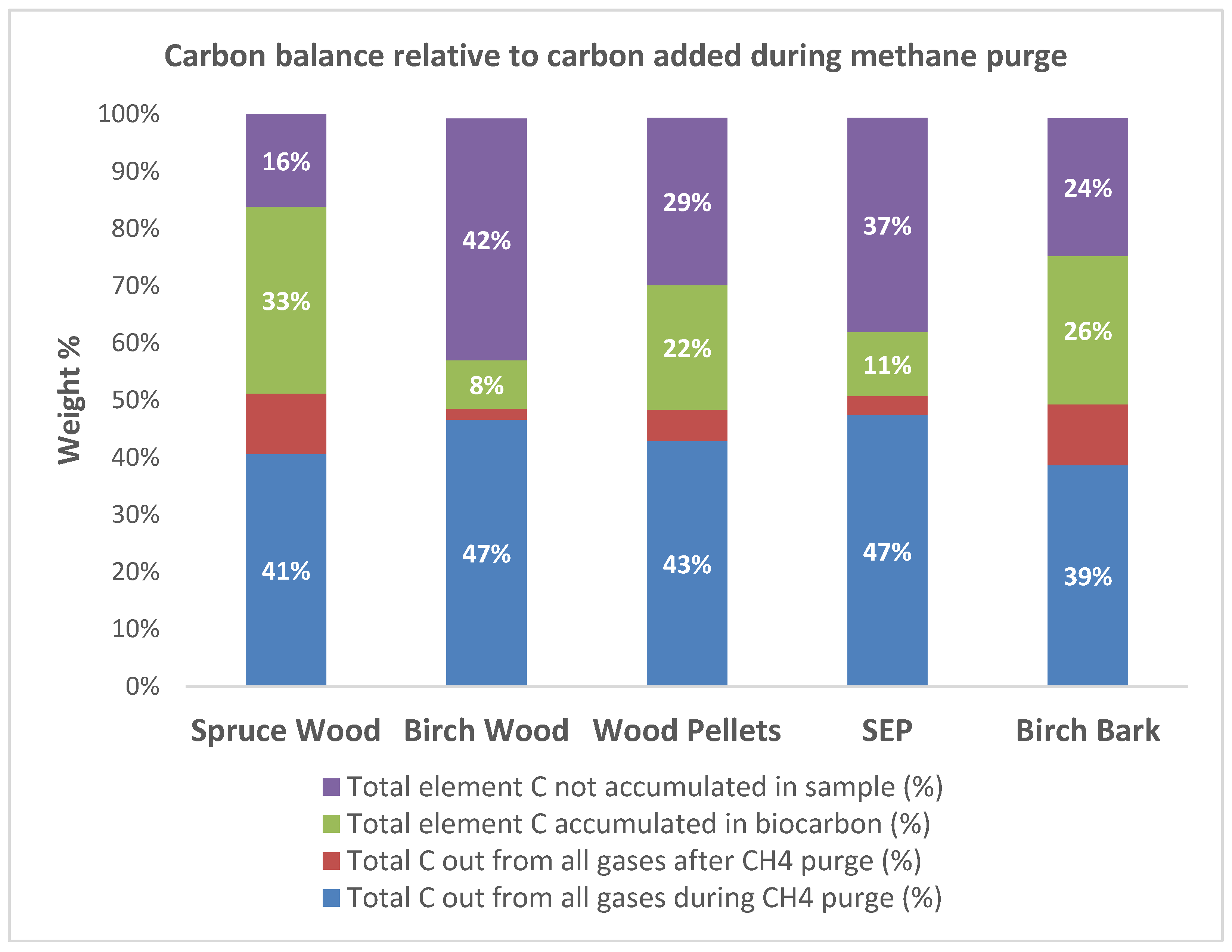
- The carbon deposition rate was derived by calculating the weight increase relative to the start weight. All the biocarbons showed high carbon deposition that increased with the reactor temperature, the methane ratio in the purge gas and the holding time. The highest deposition rate of 42.8% was achieved with birch bark, and the lowest was 13.9%, achieved with steam-explosion pellets.
- CDM experiments performed on individual samples showed that alkali metals play an important role in methane decomposition. Ninety percent of the alkali metals potassium and sodium were removed from the biocarbon after treatment at 1100 °C with a CH4/N2 ratio of 90/10 and a 90 min holding time. The alkali removal during methane decomposition also led to lower reactivity in the treated biocarbon. In fact, the potassium and sodium increase in the biocarbon yielded a higher hydrogen production rate and led to an improved CO2 reaction rate. This effect is clearly shown in Figures S2 and S6 in the Supplementary Materials.
- The surface areas of the different biocarbons also affected the hydrogen production rates. Figure S3 shows that an increase in the surface area resulted in higher hydrogen yields.
- Raman and SEM-EDS also showed a more structured carbon matrix and deposition on the particle surfaces, both of which should lead to lower reactivity. The reactivity analysis also confirmed that the CO2 reactivity was indeed lowered after the biocarbon densification. The treated SEP biocarbon had the lowest reactivity rate, which was close to that of the reference coke sample, and it can thus be considered suitable as a reducing agent in manganese production.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Heintz, I.; Mousa, E.; Ye, G. Development of Fossil-Free Technologies for the Metallurgical Industry—Swerim Pilot and Industrial Experiences. In Advances in Pyrometallurgy; Fleuriault, C., Steenkamp, J.D., Gregurek, D., White, J.F., Reynolds, Q.G., Mackey, P.J., Hockaday, S.A.C., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 55–66. [Google Scholar]
- Souza Filho, I.R.; Springer, H.; Ma, Y.; Mahajan, A.; da Silva, C.C.; Kulse, M.; Raabe, D. Green Steel at Its Crossroads: Hybrid Hydrogen-Based Reduction of Iron Ores. J. Clean. Prod. 2022, 340, 130805. [Google Scholar] [CrossRef]
- Abdul Quader, M.; Ahmed, S.; Dawal, S.Z.; Nukman, Y. Present Needs, Recent Progress and Future Trends of Energy-Efficient Ultra-Low Carbon Dioxide (CO2) Steelmaking (ULCOS) Program. Renew. Sustain. Energy Rev. 2016, 55, 537–549. [Google Scholar] [CrossRef]
- Dalaker, H.; Hovig, E.W. Hydrogen Plasma-Based Reduction of Metal Oxides. In Advances in Pyrometallurgy; Fleuriault, C., Steenkamp, J.D., Gregurek, D., White, J.F., Reynolds, Q.G., Mackey, P.J., Hockaday, S.A.C., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 85–94. [Google Scholar]
- Schanche, T.L.; Tangstad, M. Prereduction of Nchwaning Ore in CO/CO2/H2 Gas Mixtures. Minerals 2021, 11, 1097. [Google Scholar] [CrossRef]
- Osen, K.S.; Dalaker, H.; Martinez, A.M.; Gudbrandsen, H.; Wang, Z.; Kero, I. CO2 Free FeMn/Mn Production Through Molten Oxide Electrolysis. In Advances in Pyrometallurgy; Fleuriault, C., Steenkamp, J.D., Gregurek, D., White, J.F., Reynolds, Q.G., Mackey, P.J., Hockaday, S.A.C., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 267–280. [Google Scholar]
- Allanore, A. Features and Challenges of Molten Oxide Electrolytes for Metal Extraction. J. Electrochem. Soc. 2014, 162, E13. [Google Scholar] [CrossRef]
- Wiencke, J.; Lavelaine, H.; Kane, G.J.; Panteix, P.-J.; Petitjean, C.; Rapin, C. Kinetics of Iron Electrochemical Reduction into Liquid Metal at 1823 K in a Molten Oxide Electrolyte. Mater. Chem. Phys. 2018, 212, 214–223. [Google Scholar] [CrossRef]
- Myrhaug, E.H. Non-Fossil Reduction Materials in the Silicon Process-Properties and Behaviour. Ph.D. Thesis, Norges Teknisk-Naturvitenskapelige Universitet, Trondheim, Norway, 2003. Available online: https://www.osti.gov/etdeweb/servlets/purl/20559402 (accessed on 1 February 2024).
- Kaffash, H.; Surup, G.R.; Tangstad, M. Densification of Biocarbon and Its Effect on CO2 Reactivity. Processes 2021, 9, 193. [Google Scholar] [CrossRef]
- Abbas, H.F.; Wan Daud, W.M.A. Hydrogen Production by Methane Decomposition: A Review. Int. J. Hydrogen Energy 2010, 35, 1160–1190. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Chen, H.; Qi, M.; Zhang, G.; Hu, H.; Ma, X. Hydrogen Production by Catalytic Methane Decomposition: Carbon Materials as Catalysts or Catalyst Supports. Int. J. Hydrogen Energy 2017, 42, 19755–19775. [Google Scholar] [CrossRef]
- Suelves, I.; Lazaro, M.; Moliner, R.; Corbella, B.; Palacios, J. Hydrogen Production by Thermo Catalytic Decomposition of Methane on Ni-Based Catalysts: Influence of Operating Conditions on Catalyst Deactivation and Carbon Characteristics. Int. J. Hydrogen Energy 2005, 30, 1555–1567. [Google Scholar] [CrossRef]
- Amin, A.; Epling, W.; Croiset, E. Reaction and Deactivation Rates of Methane Catalytic Cracking over Nickel. Ind. Eng. Chem. Res. 2011, 50, 12460–12470. [Google Scholar] [CrossRef]
- Shah, N.; Panjala, D.; Huffman, G.P. Hydrogen Production by Catalytic Decomposition of Methane. Energy Fuels 2001, 15, 1528–1534. [Google Scholar] [CrossRef]
- Shen, W.; Wang, Y.; Shi, X.; Shah, N.; Huggins, F.; Bollineni, S.; Seehra, M.; Huffman, G. Catalytic Nonoxidative Dehydrogenation of Ethane over Fe–Ni and Ni Catalysts Supported on Mg(Al)O to Produce Hydrogen and Easily Purified Carbon Nanotubes. Energy Fuels 2007, 21, 3520–3529. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lua, A.C. Development of Metallic Nickel Nanoparticle Catalyst for the Decomposition of Methane into Hydrogen and Carbon Nanofibers. J. Phys. Chem. C 2012, 116, 26765–26775. [Google Scholar] [CrossRef]
- Moliner, R.; Suelves, I.; Lazaro, M.; Moreno, O. Thermocatalytic Decomposition of Methane over Activated Carbons: Influence of Textural Properties and Surface Chemistry. Int. J. Hydrogen Energy 2005, 30, 293–300. [Google Scholar] [CrossRef]
- Kim, M. Hydrogen Production by Catalytic Decomposition of Methane over Activated Carbons: Kinetic Study. Int. J. Hydrogen Energy 2004, 29, 187–193. [Google Scholar] [CrossRef]
- Lee, K.K.; Han, G.Y.; Yoon, K.J.; Lee, B.K. Thermocatalytic Hydrogen Production from the Methane in a Fluidized Bed with Activated Carbon Catalyst. Catal. Today 2004, 93–95, 81–86. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, H.; Li, B.; Li, W. Catalytic Decomposition of Methane over Activated Carbon. J. Anal. Appl. Pyrolysis 2005, 73, 335–341. [Google Scholar] [CrossRef]
- Abbas, H.F.; Daud, W.M.A.W. Thermocatalytic Decomposition of Methane for Hydrogen Production Using Activated Carbon Catalyst: Regeneration and Characterization Studies. Int. J. Hydrogen Energy 2009, 34, 8034–8045. [Google Scholar] [CrossRef]
- Song, Q.; Xiao, R.; Li, Y.; Shen, L. Catalytic Carbon Dioxide Reforming of Methane to Synthesis Gas over Activated Carbon Catalyst. Ind. Eng. Chem. Res. 2008, 47, 4349–4357. [Google Scholar] [CrossRef]
- Lee, E.K.; Lee, S.Y.; Han, G.Y.; Lee, B.K.; Lee, T.-J.; Jun, J.H.; Yoon, K.J. Catalytic Decomposition of Methane over Carbon Blacks for CO2-Free Hydrogen Production. Carbon 2004, 42, 2641–2648. [Google Scholar] [CrossRef]
- Muradov, N.; Smith, F.; T-Raissi, A. Catalytic Activity of Carbons for Methane Decomposition Reaction. Catal. Today 2005, 102–103, 225–233. [Google Scholar] [CrossRef]
- Suelves, I.; Pinilla, J.L.; Lázaro, M.J.; Moliner, R. Carbonaceous Materials as Catalysts for Decomposition of Methane. Chem. Eng. J. 2008, 140, 432–438. [Google Scholar] [CrossRef]
- Wei, L.; Tan, Y.; Han, Y.; Zhao, J.; Wu, J.; Zhang, D. Hydrogen Production by Methane Cracking over Different Coal Chars. Fuel 2011, 90, 3473–3479. [Google Scholar] [CrossRef]
- Wei, L.; Zhu, M.; Ma, Y.; Zhang, Z.; Zhang, D. Hydrogen Production by Methane Cracking over Xiaolongtan Lignite Chars: The Role of Mineral Matter. Fuel 2016, 183, 345–350. [Google Scholar] [CrossRef]
- Hussein, A.; Larachi, F.; Ziegler, D.; Alamdari, H. Effects of Heat Treatment and Acid Washing on Properties and Reactivity of Charcoal. Biomass Bioenergy 2016, 90, 101–113. [Google Scholar] [CrossRef]
- Wang, L.; Khalil, R.; Bakken, J.; Skreiberg, Ø. Production and Characterization of Biocarbon from Woody Biomasses Produced at 1000 °C. Fuel 2024, 370, 131667. [Google Scholar] [CrossRef]
- D1762-84R13; D07 Committee ASTM International. Test Method for Chemical Analysis of Wood Charcoal. ASTM International: West Conshohocken, PA, USA, 2021. [CrossRef]
- Maziarka, P.; Wurzer, C.; Arauzo, P.J.; Dieguez-Alonso, A.; Mašek, O.; Ronsse, F. Do You BET on Routine? The Reliability of N2 Physisorption for the Quantitative Assessment of Biochar’s Surface Area. Chem. Eng. J. 2021, 418, 129234. [Google Scholar] [CrossRef]
- Wang, L.; Olsen, M.N.P.; Moni, C.; Dieguez-Alonso, A.; de la Rosa, J.M.; Stenrød, M.; Liu, X.; Mao, L. Comparison of Properties of Biochar Produced from Different Types of Lignocellulosic Biomass by Slow Pyrolysis at 600 °C. Appl. Energy Combust. Sci. 2022, 12, 100090. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Cao, A.; Pandya, A.K.; Serhatkulu, G.K.; Weber, R.E.; Dai, H.; Thakur, J.S.; Naik, V.M.; Naik, R.; Auner, G.W.; Rabah, R.; et al. A Robust Method for Automated Background Subtraction of Tissue Fluorescence. J. Raman Spectrosc. 2007, 38, 1199–1205. [Google Scholar] [CrossRef]
- Phounglamcheik, A.; Wang, L.; Romar, H.; Kienzl, N.; Broström, M.; Ramser, K.; Skreiberg, Ø.; Umeki, K. Effects of Pyrolysis Conditions and Feedstocks on the Properties and Gasification Reactivity of Charcoal from Woodchips. Energy Fuels 2020, 34, 8353–8365. [Google Scholar] [CrossRef]
- Ishimaru, K.; Hata, T.; Bronsveld, P.; Nishizawa, T.; Imamura, Y. Characterization of Sp2- and Sp3-Bonded Carbon in Wood Charcoal. J. Wood Sci. 2007, 53, 442–448. [Google Scholar] [CrossRef]
- Sheng, C. Char Structure Characterised by Raman Spectroscopy and Its Correlations with Combustion Reactivity. Fuel 2007, 86, 2316–2324. [Google Scholar] [CrossRef]
- Lin, X.; Dong, M.; Nie, W.; Rao, G.; Lu, J. Conversion Mechanism of Organic Components during the Eucalyptus Biomass Pyrolysis Process Based on the Multispectral Association Method. Energy Fuels 2024, 38, 7929–7940. [Google Scholar] [CrossRef]
- Monsen, B.; Tangstad, M.; Solheim, I.; Syvertsen, M.; Ishak, R.; Midtgaard, H. Charcoal for manganese alloy production. In Proceedings of the Infacon XI, New Delhi, India, 18–21 February 2007; pp. 18–21. [Google Scholar]
- Zahra, A.C.A.; Anniwaer, A.; Okura, H.; Chaerusani, V.; Zhang, P.; Rizkiana, J.; Kurnia, I.; Abudula, A.; Guan, G. Addition of Torrefied Algal Biomass to Improve Land-Based Biomass Gasification for Hydrogen-Rich Gas Production. Algal Res. 2023, 74, 103236. [Google Scholar] [CrossRef]
- Brown, R.A.; Kercher, A.K.; Nguyen, T.H.; Nagle, D.C.; Ball, W.P. Production and Characterization of Synthetic Wood Chars for Use as Surrogates for Natural Sorbents. Org. Geochem. 2006, 37, 321–333. [Google Scholar] [CrossRef]
- Guizani, C.; Escudero Sanz, F.J.; Salvador, S. The Nature of the Deposited Carbon at Methane Cracking over a Nickel Loaded Wood-Char. Comptes Rendus Chim. 2016, 19, 423–432. [Google Scholar] [CrossRef]
- Kameya, Y.; Hanamura, K. Kinetic and Raman Spectroscopic Study on Catalytic Characteristics of Carbon Blacks in Methane Decomposition. Chem. Eng. J. 2011, 173, 627–635. [Google Scholar] [CrossRef]
- Muradov, N. Catalysis of Methane Decomposition over Elemental Carbon. Catal. Commun. 2001, 2, 89–94. [Google Scholar] [CrossRef]
- Yamauchi, S.; Kurimoto, Y. Raman Spectroscopic Study on Pyrolyzed Wood and Bark of Japanese Cedar: Temperature Dependence of Raman Parameters. J. Wood Sci. 2003, 49, 235–240. [Google Scholar] [CrossRef]
- Jayakumari, S.; Jahrsengene, G.; Ksiazek, M.; Ringdalen, E. Investigations on CO2 Reactivity and Thermal Strength of Carbon Sources for Ferroalloy and Silicon Production. In Proceedings of the 62nd Conference of Metallurgists, COM 2023, Toronto, ON, Canada, 21–24 August 2023; Springer Nature: Cham, Switzerland, 2023; pp. 1009–1018. [Google Scholar]
- Kaffash, H.; Tangstad, M. The Effect of Densification on Compressive Strength of Charcoal. In Proceedings of the 16th International Ferro-Alloys Congress (INFACON XVI), Virtual, 27–29 September 2021. [Google Scholar]
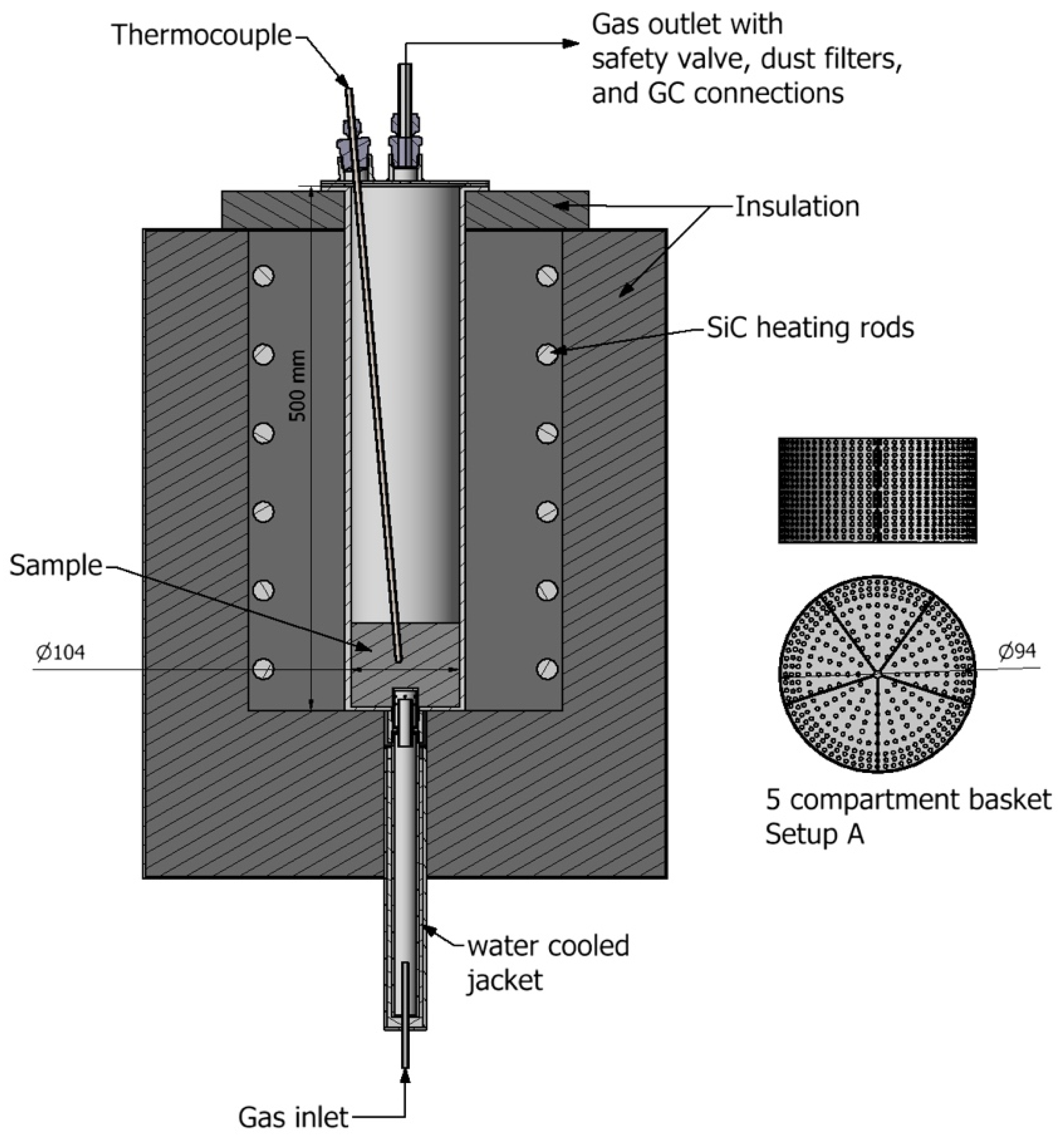
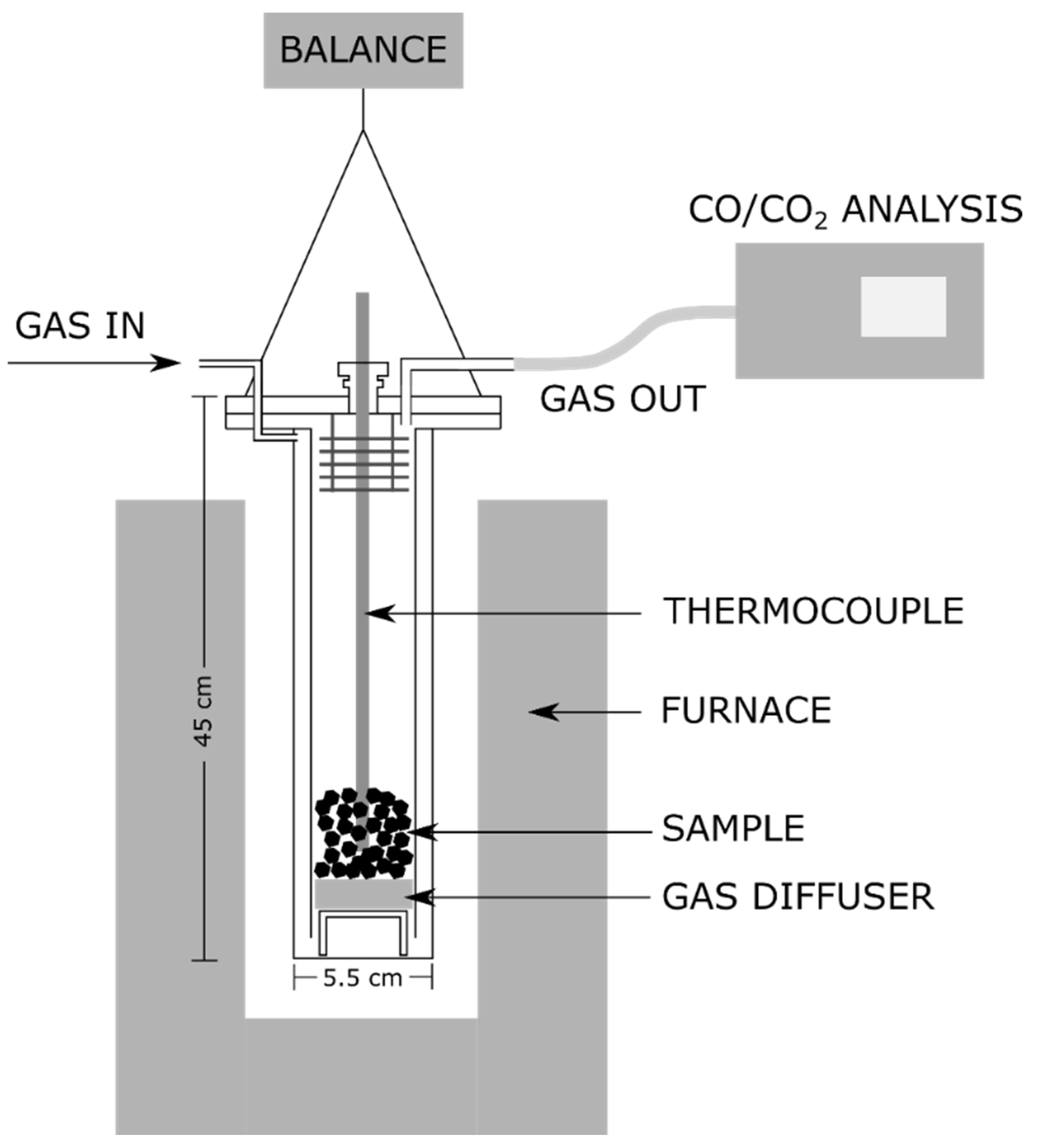


| Exp. No. | Material | Reactor T. (°C) | Holding Time (min) | Environment | Gas Analysis |
|---|---|---|---|---|---|
| 1 | All | 1000 | 30 | 90/10 CH4/Ar | No |
| 2 | All | 1000 | 90 | 90/10 CH4/Ar | No |
| 3 | All | 1100 | 30 | 90/10 CH4/Ar | No |
| 4 | All | 1100 | 90 | 45/55 CH4/Ar | No |
| 5 | All | 1100 | 90 | 90/10 CH4/Ar | No |
| 6 | All | 1100 | 90 | 0/100 CH4/Ar | No |
| 7 | Spruce Wood | 1100 | 75 | 90/10 CH4/N2 | Yes |
| 8 | Birch Wood | 1100 | 90 | 90/10 CH4/N2 | Yes |
| 9 | Wood Pellets | 1100 | 90 | 90/10 CH4/N2 | Yes |
| 10 | SEPs | 1100 | 90 | 90/10 CH4/N2 | Yes |
| 11 | Birch Bark | 1100 | 90 | 90/10 CH4/N2 | Yes |
| Feedstock | Spruce Wood | Wood Pellets | SEPs | Birch Bark |
|---|---|---|---|---|
| Start weight of biocarbon (g) | 81.7 | 80.0 | 100.0 | 82.5 |
| Volatiles released prior to CH4 purge (g) and in % relative to start weight | 0.02 (0.02%) | 0.74 (0.93%) | 0.82 (0.82%) | 0.99 (1.2%) |
| Weight prior to CH4 purge (g) | 81.7 | 79.2 | 99.2 | 81.5 |
| Weight gained (g) and in % relative to start weight | 34.36 (42.1%) | 26.48 (33.4%) | 13.8 (13.9%) | 34.91 (42.8%) |
| Total H2 out (mol) | 7.3 | 7.4 | 6.8 | 8.7 |
| Total CH4 out (mol) | 3.3 | 4.0 | 4.5 | 4.0 |
| Total C2H4 out (mol) | 0.1 | 0.1 | 0.1 | 0.1 |
| Total H2 out (mol/mol CH4 in) | 0.8 | 0.7 | 0.7 | 0.8 |
| Total H2 out (mol/kg biocarbon) | 89.9 | 92.4 | 68.2 | 105.2 |
| Total CH4 out (mol/kg biocarbon) | 40.1 | 50.6 | 44.8 | 48.4 |
| Spruce Wood | Birch Wood | Wood Pellets | SEPs | Birch Bark | |
|---|---|---|---|---|---|
| Biocarbon from pyrolysis | |||||
| Carbon | 89.42 (±0.26) | 89.67 (±0.31) | 88.99 (±0.30) | 90.23 (±0.41) | 88.10 (±0.41) |
| Hydrogen | 0.79 (±0.04) | 0.80 (±0.08) | 0.78 (±0.09) | 0.68 (±0.06) | 0.82 (±0.08) |
| Nitrogen | 0.51 (±0.007) | 0.47 (±0.004) | 0.44 (±0.005) | 0.57 (±0.002) | 0.52 (±0.002) |
| Sulphur | 0.02 (±0.001) | 0.02 (±0.001) | 0.02 (±0.001) | 0.05 (±0.001) | 0.02 (±0.001) |
| Oxygen | 9.27 | 9.04 | 9.77 | 8.47 | 10.54 |
| Treated biocarbon, at 1100 °C, 90 min, and 90/10 (CH4/N2) | |||||
| Carbon | 98.77 (±0.23) | 96.56 (±0.18) | 96.03 (±0.24) | 96.65 (±0.19) | 96.81 (±0.25) |
| Hydrogen | 0.08 (±0.01) | 0.23 (±0.02) | 0.29 (±0.02) | 0.15 (±0.02) | 0.12 (±0.02) |
| Nitrogen | 0.10 (±0.032) | 0.34 (±0.016) | 0.25 (±0.013) | 0.31 (±0.017) | 0.38 (±0.016) |
| Sulphur | 0.01 (±0.001) | 0.02 (±0.001) | 0.03 (±0.001) | 0.05 (±0.001) | 0.02 (±0.001) |
| Oxygen | 1.04 | 2.85 | 3.40 | 2.84 | 2.67 |
| Treated biocarbon, at 1000 °C, 30 min, and 90/10 (CH4/Ar) | |||||
| Carbon | 94.66 (±0.13) | 92.00 (±0.25) | 92.60 (±0.32) | 94.19 (±0.29) | 91.11 (±0.25) |
| Hydrogen | 0.31 (±0.02) | 0.52 (±0.03) | 0.44 (±0.10) | 0.45 (±0.03) | 0.64 (±0.05) |
| Nitrogen | 0.50 (±0.019) | 0.68 (±0.017) | 0.72 (±0.026) | 0.77 (±0.021) | 0.91 (±0.023) |
| Sulphur | 0.01 (±0.001) | 0.05 (±0.001) | 0.01 (±0.001) | 0.08 (±0.001) | 0.02 (±0.001) |
| Oxygen | 4.52 | 6.75 | 6.23 | 4.51 | 7.33 |
| Treated biocarbon, at 1000 °C, 90 min, and 90/10 (CH4/Ar) | |||||
| Carbon | 97.61 (±0.33) | 96.95 (±0.28) | 96.36 (±0.28) | 95.80 (±0.36) | 94.20 (±0.66) |
| Hydrogen | 0.24 (±0.054) | 0.21 (±0.033) | 0.17 (±0.038) | 0.16 (±0.063) | 0.24 (±0.05) |
| Nitrogen | 0.69 (±0.012) | 0.49 (±0.021) | 0.56 (±0.019) | 0.79 (±0.028) | 0.88 (±0.048) |
| Sulphur | 0.02 (±0.002) | 0.014 (±0.002) | 0.012 (±0.002) | 0.016 (±0.002) | 0.019 (±0.002) |
| Oxygen | 1.45 | 2.35 | 2.91 | 3.25 | 4.68 |
| Treated biocarbon, at 1100 C, 30 min, and 90/10 (CH4/Ar) | |||||
| Carbon | 95.00 (±0.41) | 95.56 (±0.36) | 95.11 (±0.48) | 95.03 (±0.45) | 94.96 (±0.19) |
| Hydrogen | 0.27 (±0.036) | 0.17 (±0.021) | 0.16 (±0.022) | 0.31 (±0.038) | 0.26 (±0.047) |
| Nitrogen | 0.18 (±0.029) | 0.30 (±0.035) | 0.27 (±0.045) | 0.34 (±0.053) | 0.31 (±0.019) |
| Sulphur | 0.01 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) |
| Oxygen | 4.54 | 3.96 | 4.45 | 4.31 | 4.45 |
| Treated biocarbon, at 1100 °C, 90 min, and 45/55 (CH4/Ar) | |||||
| Carbon | 96.67 (±0.23) | 95.68 (±0.22) | 95.36 (±0.31) | 95.19 (±0.35) | 95.16 (±0.44) |
| Hydrogen | 0.21 (±0.03) | 0.19 (±0.02) | 0.19 (±0.01) | 0.14 (±0.03) | 0.19 (±0.02) |
| Nitrogen | 0.20 (±0.02) | 0.29 (±0.04) | 0.25 (±0.04) | 0.25 (±0.05) | 0.55 (±0.02) |
| Sulphur | 0.014 (±0.001) | 0.013 (±0.001) | 0.016 (±0.001) | 0.02 (±0.001) | 0.019 (±0.001) |
| Oxygen | 2.91 | 3.84 | 4.19 | 4.40 | 4.11 |
| Treated biocarbon, at 1100 °C, 90 min, and 90/10 (CH4/Ar) | |||||
| Carbon | 96.73 (±0.13) | 95.99 (±0.15) | 95.59 (±0.18) | 95.56 (±0.19) | 95.38 (±0.19) |
| Hydrogen | 0.24 (±0.01) | 0.21 (±0.02) | 0.21 (±0.02) | 0.19 (±0.02) | 0.23 (±0.01) |
| Nitrogen | 0.27 (±0.023) | 0.26 (±0.014) | 0.20 (±0.011) | 0.18 (±0.012) | 0.24 (±0.013) |
| Sulphur | 0.05 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) | 0.02 (±0.001) |
| Oxygen | 2.71 | 3.53 | 4.00 | 4.06 | 4.13 |
| Treated biocarbon, at 1100 °C, 90 min, and 0/100 (CH4/Ar) | |||||
| Carbon | 95.53 (±0.19) | 94.01 (±0.14) | 95.97 (±0.25) | 95.23 (±0.27) | 93.89 (±0.19) |
| Hydrogen | 0.15 (±0.001) | 0.15 (±0.002) | 0.12 (±0.005) | 0.14 (±0.005) | 0.14 (±0.004) |
| Nitrogen | 0.55 (±0.009) | 0.68 (±0.004) | 0.63 (±0.002) | 0.47 (±0.002) | 0.44 (±0.001) |
| Sulphur | 0.01 (±0.29) | 0.09 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) | 0.01 (±0.001) |
| Oxygen | 3.77 | 5.07 | 3.26 | 4.15 | 5.52 |
| Ash Analysis | Spruce Wood | Birch Wood | Wood Pellets | SEPs | Birch Bark |
|---|---|---|---|---|---|
| Biocarbon from pyrolysis | |||||
| Calcium | 3620 | 4151 | 2890 | 3280 | 16,549 |
| Potassium | 2770 | 3595 | 1797 | 1396 | 8048 |
| Phosphorus | 291 | 240 | 272 | 142 | 957 |
| Silicon | 134 | 169 | 122 | 123 | 340 |
| Sodium | 1861 | 1885 | 1699 | 1047 | 6831 |
| Sulphur | 158 | 175 | 98 | 140 | 637 |
| Magnesium | 381 | 505 | 352 | 262 | 1285 |
| Manganese | 352 | 304 | 346 | 247 | 859 |
| Aluminum | 212 | 270 | 142 | 125 | 353 |
| Iron | 224 | 330 | 216 | 139 | 460 |
| Copper | 3 | 5 | 1 | 4 | 5 |
| Zinc | 9 | 11 | 4 | 4 | 5 |
| Barium | 58 | 67 | 48 | 79 | 354 |
| Titanium | 7 | 21 | 11 | 4 | 21 |
| Treated biocarbon, at 1100 °C, 90 min, and 90/10 (CH4/N2) | |||||
| Calcium | 2550 | 3706 | 2350 | 3260 | 14,780 |
| Potassium | 340 | 219 | 199 | 170 | 4578 |
| Phosphorus | 150 | 143 | 138 | 89 | 357 |
| Silicon | 119 | 134 | 154 | 110 | 200 |
| Sodium | 106 | 202 | 135 | 106 | 180 |
| Sulphur | 98 | 162 | 113 | 70 | 287 |
| Magnesium | 273 | 372 | 408 | 390 | 1174 |
| Manganese | 151 | 259 | 260 | 384 | 406 |
| Aluminum | 64 | 140 | 88 | 85 | 156 |
| Iron | 198 | 441 | 182 | 102 | 278 |
| Copper | 3 | 2 | 3 | 2 | 6 |
| Zinc | 4 | 7 | 5 | 4 | 9 |
| Barium | 36 | 55 | 30 | 69 | 205 |
| Titanium | 3 | 8 | 7 | 1 | 13 |
| Treated biocarbon, at 1000 °C, 30 min, and 90/10 (CH4/Ar) | |||||
| Calcium | 3150 | 3110 | 2620 | 3030 | 16,280 |
| Potassium | 1060 | 840 | 350 | 850 | 6440 |
| Phosphorus | 210 | 240 | 206 | 200 | 1320 |
| Silicon | 108 | 134 | 139 | 109 | 441 |
| Sodium | 131 | 227 | 147 | 191 | 374 |
| Sulphur | 80 | 36 | 45 | 120 | 280 |
| Magnesium | 369 | 412 | 719 | 407 | 1196 |
| Manganese | 226 | 291 | 326 | 405 | 772 |
| Aluminum | 74 | 42 | 32 | 76 | 86 |
| Iron | 149 | 459 | 208 | 108 | 392 |
| Copper | 6 | 6 | 5 | 136 | 93 |
| Zinc | 5 | 4 | 9 | 4 | 4 |
| Barium | 53 | 68 | 33 | 71 | 308 |
| Titanium | 10 | 10 | 1 | 6 | 9 |
| Treated biocarbon, at 1100 °C, 90 min, and 90/10 (CH4/Ar) | |||||
| Calcium | 3211 | 2182 | 2819 | 2618 | 11,285 |
| Potassium | 123 | 164 | 218 | 200 | 143 |
| Phosphorus | 27 | 421 | 24 | 128 | 1138 |
| Silicon | 140 | 168 | 173 | 135 | 402 |
| Sodium | 151 | 182 | 100 | 64 | 160 |
| Sulphur | 112 | 183 | 128 | 156 | 224 |
| Magnesium | 340 | 973 | 357 | 383 | 1165 |
| Manganese | 94 | 291 | 87 | 361 | 446 |
| Aluminum | 59 | 109 | 97 | 81 | 172 |
| Iron | 68 | 466 | 173 | 284 | 250 |
| Copper | 2 | 5 | 2 | 14 | 5 |
| Zinc | 4 | 9 | 5 | 34 | 6 |
| Barium | 55 | 36 | 70 | 63 | 197 |
| Titanium | 5 | 10 | 5 | 21 | 13 |
| Treated biocarbon, at 1100 °C, 90 min, and 0/100 (CH4/Ar) | |||||
| Calcium | 3079 | 2780 | 3710 | 3031 | 14,770 |
| Potassium | 1456 | 1630 | 1410 | 1382 | 6800 |
| Phosphorus | 189 | 170 | 123 | 220 | 620 |
| Silicon | 156 | 187 | 181 | 143 | 413 |
| Sodium | 1045 | 1175 | 691 | 506 | 2288 |
| Sulphur | 118 | 100 | 60 | 80 | 210 |
| Magnesium | 282 | 431 | 341 | 260 | 1122 |
| Manganese | 312 | 266 | 297 | 376 | 989 |
| Aluminum | 95 | 85 | 37 | 65 | 311 |
| Iron | 49 | 138 | 74 | 178 | 241 |
| Copper | 7 | 9 | 6 | 6 | 12 |
| Zinc | 4 | 4 | 4 | 4 | 4 |
| Barium | 92 | 47 | 67 | 84 | 392 |
| Titanium | 1 | 13 | 1 | 1 | 21 |
| Physico-Chemical Property | Spruce Wood | Birch Wood | Wood Pellets | SEPs | Birch Bark |
|---|---|---|---|---|---|
| Biocarbon from pyrolysis | |||||
| Surface area (N2) (m2 g−1) | 204 | 223 | 150 | 105 | 279 |
| Density (g cm−3) | 1.97 | 1.99 | 2.04 | 2.02 | 1.70 |
| Treated biocarbon | |||||
| Surface area (N2) (m2 g−1) | 167 (18%) | 148 (34%) | 133 (11%) | 88 (16%) | 203 (27%) |
| Density (g cm−3) | 1.79 (9%) | 1.81 (9%) | 1.69 (17%) | 1.97 (3%) | 1.62 (5%) |
| Parameter | Spruce Wood | Birch Wood | Wood Pellets | SEPs | Birch Bark |
|---|---|---|---|---|---|
| ID/IG | 1.76 | 1.37 | 1.25 | 1.16 | 0.99 |
| AD/AG | 3.08 | 2.35 | 1.29 | 1.51 | 1.26 |
| AD/Atotal | 0.54 | 0.46 | 0.37 | 0.32 | 0.33 |
| FWHMD (cm−1) | 159.3 | 127.5 | 120.2 | 101.2 | 104.3 |
| FWHMG (cm−1) | 95.2 | 102.6 | 104.2 | 98.7 | 96.4 |
| CO2-Reactivity | Start wt. (g) | End wt. (g) | Wt. Loss (%) | Overall RR 1 | 1–2 min RR 2 | 2–4 min. RR 3 |
|---|---|---|---|---|---|---|
| Non-treated biocarbons | ||||||
| SW | 20 | 17.9 | 10.6 | 9.46 | 8.48 | - |
| BW | 19 | 17.1 | 14.5 | 9.95 | 9.29 | 7.72 |
| WP | 20 | 17.7 | 11.7 | 6.1 | 6.8 | 4.56 |
| SEP | 20 | 18.1 | 9.5 | 5.05 | 5.17 | 4.02 |
| BB | 20 | 17.2 | 14.2 | 11.37 | 9.22 | - |
| Treated biocarbons | ||||||
| SW | 20 | 18.9 | 5.9 | 3.88 | 5.24 | 2.59 |
| BW | 20 | 18.9 | 5.6 | 4.37 | 4.79 | 3.32 |
| WP | 20 | 17.8 | 11.1 | 4.83 | 5.86 | 3.49 |
| SEP | 20 | 18.9 | 5.5 | 2.62 | 4.45 | 2.53 |
| BB | 20 | 19.3 | 3.7 | 3.81 | 5.16 | 2.66 |
| Reference (coke) | ||||||
| 20 | 17.8 | 1.1 | 2.66 | 4.07 | 1.85 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, R.A.; Jayakumari, S.; Dalaker, H.; Wang, L.; Tetlie, P.; Skreiberg, Ø. Catalytic Methane Decomposition for the Simultaneous Production of Hydrogen and Low-Reactivity Biocarbon for the Metallurgic Industry. Energies 2025, 18, 558. https://doi.org/10.3390/en18030558
Khalil RA, Jayakumari S, Dalaker H, Wang L, Tetlie P, Skreiberg Ø. Catalytic Methane Decomposition for the Simultaneous Production of Hydrogen and Low-Reactivity Biocarbon for the Metallurgic Industry. Energies. 2025; 18(3):558. https://doi.org/10.3390/en18030558
Chicago/Turabian StyleKhalil, Roger A., Sethulakshmy Jayakumari, Halvor Dalaker, Liang Wang, Pål Tetlie, and Øyvind Skreiberg. 2025. "Catalytic Methane Decomposition for the Simultaneous Production of Hydrogen and Low-Reactivity Biocarbon for the Metallurgic Industry" Energies 18, no. 3: 558. https://doi.org/10.3390/en18030558
APA StyleKhalil, R. A., Jayakumari, S., Dalaker, H., Wang, L., Tetlie, P., & Skreiberg, Ø. (2025). Catalytic Methane Decomposition for the Simultaneous Production of Hydrogen and Low-Reactivity Biocarbon for the Metallurgic Industry. Energies, 18(3), 558. https://doi.org/10.3390/en18030558