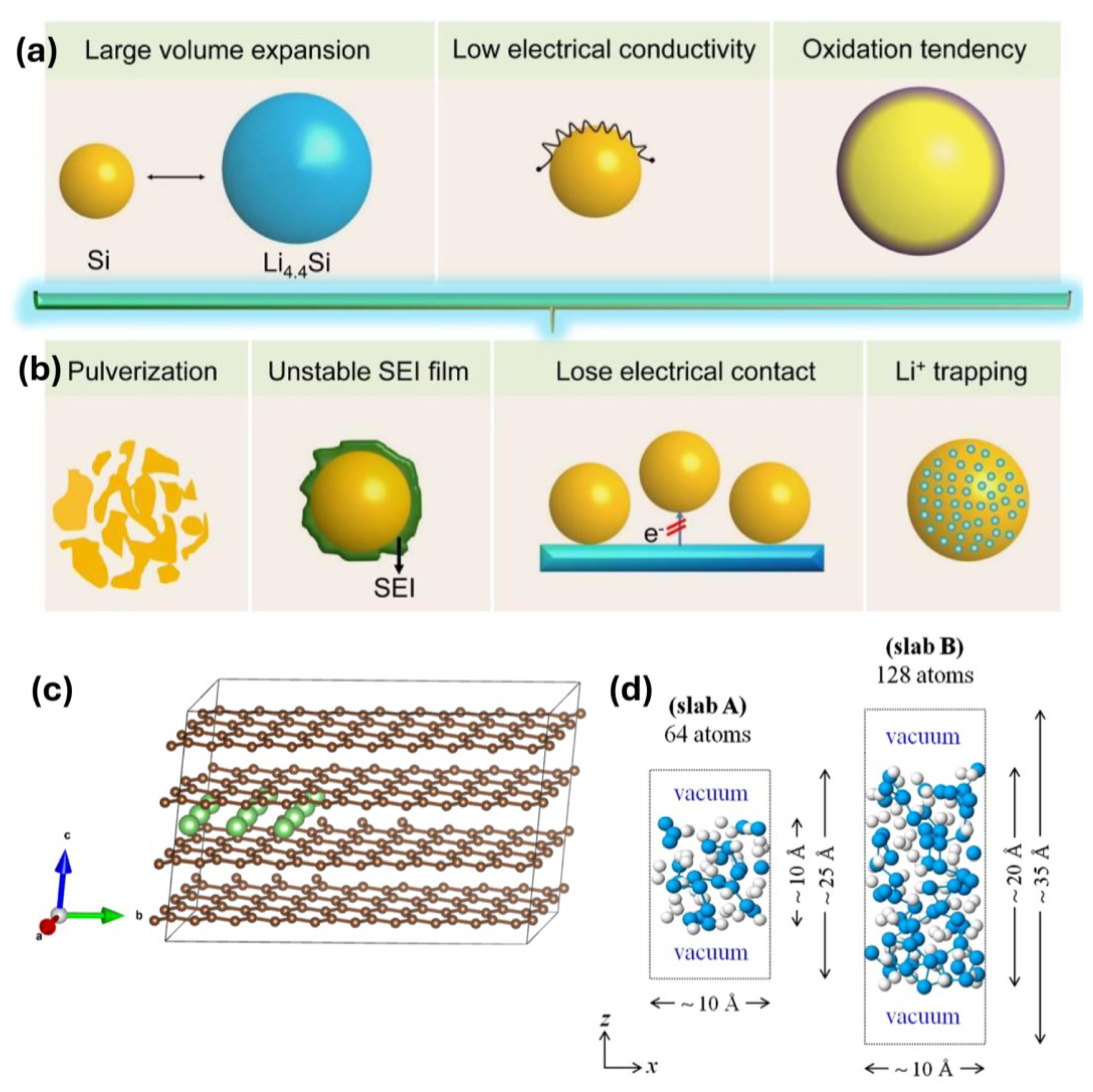
2.1. Carbon Materials
Carbon coatings have been applied to anodes and cathodes fabricated using materials such as lithium transition metal oxides (for example, LFP cathodes and silicon anodes) to improve the performance of LIBs. Carbon is an effective coating material owing to its high electrical conductivity, good elasticity, abundance, easy preparation, and low cost [
41]. Three factors determine the extent to which the performance of silicon anodes can be improved using carbon coatings. (1) The degree of order or disorder of the coated carbon, which is related to the crystallinity of the carbon coating layer, affects its performance. Systems with crystalline carbon coatings have achieved improved CE and reduced SEI layer thickness owing to their mechanical strength and flexibility; however, the degree of suppression of the volume expansion of silicon during lithiation is less than that achieved using amorphous carbon. Amorphous coatings have been used to inhibit the volume expansion of silicon during lithiation; however, they are not suitable for commercialization owing to their low structural stability and low conductivity. (2) The thickness of the carbon coating affects its performance in silicon anodes. Although the thickness for achieving optimal performance depends on the type of carbon that is coated, the thickness influences not only the capacity retention and initial Coulombic efficiency (ICE) but also the structural properties of the coated carbon layer. (3) The structure of the carbon coating (yolk–shell or core–shell) also influences its performance. Essentially, the stress generated by silicon particles during volume expansion can be relieved by adjusting the structure to provide a void space. Therefore, these three factors must be considered when optimizing the performance of carbon coatings. In the early 21st century, Yoshio et al. demonstrated the advantages and limitations of carbon-coated silicon as an anode material. Moreover, the carbon-coated silicon was compared with the original silicon, and the results revealed an improvement in electrochemical performance [
45]. However, capacity fading with cycling was observed in both the pristine and carbon-coated specimens of silicon; notably, Dimov et al. explored the underlying mechanism [
33]. Early studies suggested that the size of silicon particles for carbon coating had to be controlled to nanometer levels [
46], leading to research on the use of more optimal carbon coatings. Recent studies on the application of these appropriate carbon coatings to silicon nanoparticles have focused on the method for coating carbon and the atomic arrangement of the coated carbon, suggesting that the application varies with the type of carbon [
47].
The characteristics of the carbon coating vary with the crystallinity of the coated carbon. Crystalline carbon exhibits high mechanical strength and high electrical conductivity, enabling the electrode to exhibit rapid kinetics and good cycling performance. Whereas amorphous carbon exhibits high initial irreversible capacity owing to the lithium-ion trapping in defected parts [
48]. Meanwhile, diverse carbon substances such as graphene, amorphous carbon, nanocarbon fibers, and carbon nanotubes have been explored as carbon coating materials for silicon anodes [
49,
50]. In particular, Wang et al. used graphene as the carbon coating material [
51]. They found that coating silicon by traditional mechanical mixing not only inhibited the improvement in the electrical conductivity of the nano-silicon, owing to the nonuniform dispersion of silicon particles on graphene sheets and weak bonding between silicon particles and graphene sheets, but was also ineffective in suppressing the volume expansion owing to straightforward peel-off between the silicon particles and graphene sheets. To overcome this drawback, a three-dimensional (3D) core–shell structure was designed to effectively suppress the volume expansion and improve electrical conductivity. Therefore, the authors fabricated the 3D core–shell structure of Si@Graphene composites using the catalytic growth of graphene, coating the surface of silicon particles using nickel as a catalyst. The oxide layer on the surface of ~50 nm-sized silicon particles (0.5 g) uniformly dispersed in the mixed solvents was removed using ethanol (35 mL). Nickel was deposited on the surface of treated silicon nanoparticles at 50 ℃ for 10 min, following which the Ni-coated silicon was dispersed into a solution of triethylene glycol (38 mL) and aqueous NaOH (0.25 mL) with stirring at 185 ℃ for 14 h, which yielded carbon-incorporated Ni-coated silicon. Three-dimensional Si@Graphene composites were obtained by heating the preceding material at 450 ℃ for 60 min in an Ar gas atmosphere and removing the Ni layer by etching for several hours (
Figure 2a). Consequently, a void space was created between the graphene coating layer and silicon particles, with the coating layer present in a 2–5 nm wrinkly layer. In the galvanostatic charge/discharge profiles (
Figure 2b,c), Si@Graphene showed longer plateaus than those of pure silicon, suggesting that the ICE was low because of more side reactions between the graphene layer and the anode materials. Si@Graphene exhibited a greater initial irreversible capacity loss than that of pure silicon; however, it showed a higher CE and superior rate capability compared with those of pure silicon because it suppressed volume expansion to a greater extent and showed improved cycling stability by forming a more stable SEI layer. This change was because the graphene coating layer on the silicon nanoparticles inhibited continuous SEI formation. Additionally, cathodic peaks were observed in the profiles of both Si@Graphene and pure silicon at 1.0–1.2 V, with those of pure silicon being more intense (
Figure 2d,e); these were related to the irreversible reaction between the electrolyte and electrode surface, implying that more side reactions occurred with pure silicon than those with Si@Graphene. Furthermore, a peak was observed at 0.3–1.1 V for Si@Graphene, which was invisible in the case of silicon, indicating an additional irreversible reaction. This change caused more initial Li
+ consumption than in the scenario with pure silicon; however, the degree of irreversible Li
+ consumption at 0.01–0.2 V was considerably less in Si@Graphene than in pure silicon, suggesting that the graphene layer helped enhance the stability of the surface/interface of the electrode. Moreover, pure silicon cracked to a greater extent than that of the Si@Graphene composite after 50 cycles, and the thickness of the pure silicon electrode increased from 11.5 µm to 41.3 µm during lithiation, whereas that of Si@Graphene only increased from 11.1 µm to 28.9 µm. Additionally, the graphene coating layer caused the initial irreversible capacity loss of the silicon anode; however, it improved the interfacial and surface characteristics of the electrode and contributed to cycling stability [
52]. Kim et al. reported a study in which a graphene layer was deposited on the surface of nano-silicon. They found that the surface characteristics varied with the method used for synthesizing silicon, and accordingly, the morphology of the carbon coating formed by CVD also changed. The authors prepared a sample (NAT-Si) in which the oxide layer was removed by etching with HF and synthesized another sample (TH-Si) in which a thin, dense oxide layer was formed by thermal treatment on a 100 nm-sized silicon nanoparticle. Different structures were formed when carbon was deposited on the two different silicon samples (NAT-Si and TH-Si) through low-pressure CVD at 900 ℃. The surface of NAT-Si, which had an oxygen-poor surface layer on the silicon particles, was coated with disordered graphite, whereas the surface of TH-Si, which had an oxygen-rich surface layer on the silicon particles, was coated with ordered graphene, indicating that the thermal treatment determined the structure of the coated carbon, which subsequently affected the electrode performance. Electrochemical cycling tests were conducted on silicon half-cells synthesized using NAT-Si and TH-Si, and the results indicated that 1300 mAhg
−1, or 75.7% of the initial capacity, was maintained after 300 cycles for TH-Si with the ordered graphene coating, with the value being only 600 mAhg
−1, or 35.7% of the initial capacity, for NAT-Si with the disordered graphite coating. An analysis of this phenomenon by cryogenic electron microscopy suggested that the ordered-graphene-coated silicon withstood the mechanical strain induced by the large volume expansion, whereas the disordered-graphite-coated silicon was continuously exposed to the electrolyte and allowed the SEI layer to become thicker as it collapsed [
53]. Li et al. reported a study on a well-designed cage-type coating on crystalline silicon microparticles (SIMPs) and explored the advantages of the graphene coating over amorphous carbon coatings. This investigation targeted the volume expansion induced during the lithiation of crystalline silicon, during which it exhibited the highly anisotropic property of crystallographical expansion in certain directions. For this reason, the coating on the SIMPs was designed to withstand the stress caused by volume expansion in a specific direction. However, the amorphous carbon coating, particularly the porous layer, readily cracked and collapsed because it could not withstand the stress in a certain direction owing to its inherent abundant surface defects and low crystallinity, attributed to low conductivity and low structural stability [
48,
54]. By contrast, the coating with crystalline carbon (graphene in the cited study) could endure the stress owing to its mechanical strength and flexibility, resulting in an improved CE and a reduced SEI-layer thickness [
48,
55].
To improve the performance of silicon anodes using coatings, research has been conducted not only on coating materials but also on the suitable thickness of the carbon coating layer. Luo et al. reported a study on the use of Si@C core–shell nanoparticles to control the thickness of the coating layer at 2–25 nm and determine an appropriate thickness. In this study, amorphous silicon nanoparticles with a diameter of 80 nm were selected, and a resorcinol–formaldehyde (RF) resin layer was deposited by template sol–gel coating method and then carbonized to adjust the thickness of the coating layer (
Figure 3a). A thicker carbon coating layer with an improved thickness was obtained as the amount of resorcinol increased; for instance, the use of 0.02, 0.14, and 0.70 g of resorcinol resulted in carbon coatings with thicknesses of 2(Si@2C), 5(Si@5C), and 15(Si@15C) nm, respectively. Coatings of approximately 2, 5, 10, 15, and 25 nm thick were prepared; moreover, the carbon in the coated layer was amorphous and the silicon particles were crystallized. (Samples in
Figure 3 are denoted Si@xC, where x represents the thickness of the coating layer). The measured ICE of pristine silicon was remarkably lower (72.6%) than that of Si@5C and Si@15C samples (85.8% and 74.9%, respectively). Furthermore, the capacity retention after 200 cycles was measured to be 21%, 66%, and 69% for Si@5c, Si@10c, and Si@15c, respectively. Capacity retention was facilitated when the thickness of the coating increased by 5–15 nm, whereas high ICE was obtained when the thickness of the coating decreased. Among the prepared samples, optimal cycling performance was achieved by the specimen with a thickness of 10 nm (
Figure 3b). Electrochemical analysis indicated that the 10 nm-thick coating exhibited a high CE (>99.5%; 50th cycle to 500th cycle;
Figure 3c) and a considerably low charge transfer resistance (
Rct), which helped in achieving high rate performance and forming a stable SEI layer. Therefore, as seen in
Figure 3d, Si@10C was considered the optimal sample (high capacities of 2864, 2500, 2231, 1813, and 1209 mAh g
−1 at a current density of 0.28, 1.4, 5.6, 11.2, and 16.8 Ag
−1, respectively). Also, the capacity retention was confirmed to be superior to pristine Si when the current density increased from 0.28 to 16.8 Ag
−1. Meanwhile, the rate performance of the sample with a 15 nm coating was remarkably poor, which indicates that a too-thick coating layer acts as a source of resistance, hence causing low rate capacity at high current. A thicker layer is generally effective in mitigating the volume expansion as it enhances the performance; however, excessively thick coatings can act as a source of resistance to the movement of Li ions and inhibit the rate performance (
Figure 3d) [
56]. Recently, Qi et al. reported a study on the use of CVD to determine the appropriate thickness of the coating layer. They prepared 100 nm-sized carbon-coated silicon nanoparticles (Si@C) by reacting silicon nanoparticles with C
3H
6 gas in a vertical quartz reactor at 680 °C. Moreover, they controlled the thickness of the coating by adjusting the deposition period (10, 15, 25, 40, and 60 min, which resulted in 1, 2, 3, 4, and 5 nm-thick coatings, respectively). As shown in
Figure 3e, optimal cycling performance and a specific capacity retention of 1759 mAhg
−1 at 0.2 Ag
−1 after 500 cycles were exhibited by silicon with a carbon coating thickness of 2 nm. The 1 nm coating did not effectively prevent the silicon from being exposed to the electrolyte, whereas the 5 nm nonuniform coating hindered the insertion of Li ions into the bulk phase of Si@C. Furthermore, a porous carbon coating was formed at a specific thickness (15 nm), and the coated layer likely acted as a source of resistance when the coating was thicker than 5 nm and nonuniform. The electrochemical performance of the samples depended on the thickness of the coating, owing to differences in specific surface area and electrical conductivity (
Figure 3f). The electrical conductivity increased when the thickness was increased from 1 to 3 nm and then decreased thereafter, suggesting that the electron transfer resistance increased and the electrical conductivity decreased as the amount of isotropic carbon with more defects and disordered orientation increased [
49]. And the dramatically improved specific surface area was confirmed in a sample with a 15 nm-thick coating layer, which indicates that the porous carbon coating was formed when the thickness of the coating layer exceeded 15 nm (
Figure 3f). These studies underscore the importance of studying the thickness of the carbon coating because it affects the surface characteristics of the coating, as well as its porosity, ICE, and capacity retention, and the degree to which the volume expansion of silicon is suppressed; even these characteristics may vary depending on the experimental surroundings and conditions [
49,
56].
The efficiency of carbon coatings has also been improved by modifying the coating structure to achieve a yolk–shell coating structure. In contrast to the basic coating method (core–shell structure), the yolk–shell structure forms a gap between the coated surface and silicon particles [
57]. The yolk–shell structure contains a carbon coating, void, and silicon nanoparticles (
Figure 4a), thereby providing sufficient space for expansion and contraction during charging and discharging to protect the coating layer and silicon particles, overcoming problems faced by the core–shell structure, such as the cracking of the silicon particles and coating layer. These structural characteristics can improve the durability of the composite and the performance of the electrode [
58,
59]. Recently, Xie et al. conducted the facile synthesis of yolk–shell-structured Si@void@C nanoparticles. Approximately 80 nm-sized silicon particles were dispersed in a mixed solution of deionized water and ethanol by ultrasonication for 1 h, and the system was stirred with the addition of tetraethoxysilane to obtain Si@SiO
2. The resulting nanoparticles were ultrasonicated with cetyltrimethylammonium bromide, and the mixture was stirred for 30 min following the addition of ethanol, resorcinol, and ammonia. The resulting mixture was stirred for 6 h after adding formaldehyde, yielding Si@SiO
2@RF nanoparticles. Finally, Si@void@C particles were obtained by removing the layer of SiO
2 using an HF solution and then used to synthesize Si@void@C/CNFs (
Figure 4b,c). Cycling performance analysis of Si/CNFs, Si@C/CNFs, and Si@void@C/CNFs at a current density of 100 mAg
−1 for 100 cycles indicated that the ICE of Si/CNFs and Si@C/CNFs was 79.1% and 78.3%, respectively, owing to the creation of an amorphous carbon coating; moreover, the corresponding capacity retention values were extremely low (9.6% and 20%). The Si@void@C/CNF composite electrode retained remarkable reversible capacity above 627.5 mAhg
−1 (capacity retention of 69.3%) even after 100 cycles (
Figure 4d) because the void effectively acted as a buffer against the substantial volumetric changes; hence, fewer cracks were generated on the coating layer owing to the extra space provided for the volume expansion of silicon particles. Overall, the presence or absence of the void region—created by structurally altering the carbon coating on the silicon anode—affected the electrochemical performance of silicon-based LIBs.
In many studies, core–shell- or yolk–shell-structured carbon coatings have resulted in improved electrical conductivity and suppression of volume expansion; however, these advances remain insufficient for commercialization. Carbon coating on silicon and graphite used as active materials are more advantageous for commercialization than carbon coating on silicon as an active material, in terms of cycle stability and capacity retention. Hence, a method involving blending graphite and silicon and then coating with carbon is frequently being used to facilitate commercialization [
47,
58,
59,
60,
61]. Blending with graphite helps provide lubrication to prevent the grinding of the active material particles, leading to improved cycle performance [
62,
63]. However, this scheme hinders the advantages of silicon that aid in achieving high capacity. Additionally, carbon coating processes such as carbonization and carbon decomposition require high temperatures (>700 ℃) [
64]. Therefore, research on alternative coating materials is necessary for expanding the use of silicon anodes.
2.2. Inorganic Materials
Compared with carbon coatings, inorganic coatings can be more stable and suppress the volume expansion of silicon particles to a greater extent [
46]. Inorganic materials used in silicon anodes include oxide-based materials, nitride-based materials, and metallic materials (including metal oxides). The improved performance of silicon electrodes obtained by oxide coating and metal coating is owing to the prevention of the peeling phenomenon between the current collector and anode, which averts rapid capacity loss due to contact with the electrolyte and helps achieve rate-speed performance through the improvement in conductivity. To create uniform metal coatings, a process involving silicon particle etching has been used in many studies; this process varies with the change in the redox potential depending on the weight of the metal [
65]. And the high cost of metals is also a limitation of the application of metal materials on Si-based anodes [
66]. Additionally, the following factors affect the performance of metal coatings: (1) the presence of a uniform and stable coating, (2) the porosity of the metal coating layer, and (3) the thickness of the metal coating layer. Typically, metallic coatings with an amorphous structure exhibit greater elastic deformation than coatings with crystalline/bulk structures [
67]. Moreover, metal oxides can outperform metals as coating materials. The metal oxide layer acts as a nonconductive layer, unlike some carbon or metal coating layers that are conductive. Furthermore, the metal oxide coating layer not only acts as an artificial SEI layer (thereby suppressing electrolyte decomposition) but also increases the mechanical strength, thereby effectively suppressing the volume expansion of silicon. Additionally, inactive coatings are used in organic coatings because direct electrode–electrolyte contact is possible, and they are typically synthesized with a nanometer-scale porous structure for rapid lithium-ion intercalation. However, the low electrical conductivity could be a disadvantage of metal oxide coating materials. In particular, SiO
2 coating materials among inorganic coatings are promising owing to their straightforward synthesis. When the coating layer in the metal oxide layer becomes too thick, it may impede the flow of Li ions and exhibit the detrimental pore toughness effect [
68]. Nitride-based products have not been extensively used; nevertheless, a TiN coating has been discussed herein as a representative example. TiO
2 can outperform metal coatings; however, it is generally synthesized by changing the coating layer to TiN by processing it in TiO
2, which is difficult to commercialize given its complexity.
Metals exhibit high conductivity, ductility, and mechanical strength, in addition to small volume expansion during charging/discharging, enabling the formation of a surface protective layer with silicon alloys and facilitating electron transport, thereby reducing the irreversible capacity loss of silicon and improving its electrochemical performance [
69]. Therefore, many inert and active materials (without and with lithium intercalation activity, respectively) such as Cu, Fe, Ni, Ag, and Ge have been extensively studied [
65,
69,
70,
71,
72,
73]. The electrical contact between active materials increases when these metal materials are coated on silicon particles; representative studies conducted in this regard are discussed in [
74]. Among metal coating layers, copper is typically used to coat silicon anodes. The results of high-resolution analysis and in situ electrochemical Raman spectroscopy conducted on copper coatings on silicon anodes indicated that the charge transfer kinetics were enhanced owing to the reduction in
Rct, and the charge storage capacity was increased owing to the increased reversibility; moreover, the tolerance against the volume expansion of silicon was increased during cycling. Murugesan et al. synthesized copper-coated silicon using a low-temperature solution-based polyol method with colloidal hydrogenated amorphous silicon (a-Si:H) and a solution of copper acetate in ethylene glycol (
Figure 5a). Pretreated (colloidal hydrogenated) silicon particles were used because the content of hydrogen in the a-Si:H particles facilitated the deposition of copper particles in terms of determining the amount of copper coated on the a-Si:H surface. At a current of 100 mAg
−1, the Cu-coated a-Si:H particles exhibited a remarkably higher specific storage capacity (600 mAhg
−1) than that of pure a-Si:H (84 mAhg
−1) and graphite anodes (372 mAhg
−1) (
Figure 5b). Additionally, the specific capacity of the copper-coated a-Si:H particles increased during cycling at currents of 70–100 mAg
−1. The poor specific capacity of pristine a-Si:H was attributed to its poor conductivity; in contrast, the copper layer provided enhanced electrochemical properties (specific storage capacity), thereby preventing solvent decomposition and bestowing mechanical integrity and substantial electrical wiring functionality [
74]. Kim et al. suggested that these outcomes were due to the high conductivity of copper (5.96 × 10
5 Scm
−1) [
70]. They conducted the annealing of electrodeposited copper on the surface of silicon at high temperatures, forming the Cu
3Si alloy phase with superior long-term cyclability. They achieved a uniform, robust deposition of copper on the surface of silicon and used etching silicon powders with surface roughness. The electroless deposition was performed by immersing the etched silicon in a solution containing copper sulfate, followed by the annealing of the copper-deposited silicon powder at 400–800 °C for 9 h under an Ar atmosphere. The benefits of the etching were revealed by field-emission SEM images (FE-SEM;
Figure 5), which indicated that the surface of the deposited copper differed from that of the bare and etched silicon particles. In the case of the bare silicon particles (10 μm), the amount of copper deposited was not sufficient and the copper detached easily during the washing process; in contrast, the etched silicon, which exhibited high-density tips on the surface, facilitated uniform copper deposition owing to its large surface area and sharp tips (
Figure 5a–d). Additionally, the cycling performance of the sample annealed at 400 ℃ was superior to that of bare silicon, although copper did not alloy with lithium; thus, the charge capacity in the first cycle was lower. Notably, despite the presence of inactive copper in the copper-deposited samples (both annealed and non-annealed), the discharge capacity was higher than that of bare silicon owing to the copper-driven creation of a conductive layer on the surface of the silicon particles, which provided an electron transfer channel even during expansion and contraction (that is, the charging and discharging of the silicon anode). However, the silicon anode was affected not only by the conductivity of copper but also by the use or non-use of annealing, with improved cyclability confirmed for the annealed sample (after copper deposition) despite its conductivity being lower than that of the copper-deposited sample (without annealing). This difference was attributed to the formation of the Cu
3Si alloy phase by some of the copper particles on the silicon, owing to which copper remained on the surface of the silicon even after cycling, whereas the sample involving copper deposition without annealing fragmented only after three cycles [
75].
Sethuraman et al. reported the improved cyclability of silicon thin films coated with porous copper and suggested that the amorphous copper gained elasticity and was able to overcome the short-lived cycling due to delamination between the current collector and active material. The porous copper coating was bound to the entire electrode and prevented the exposure of fresh silicon to the electrolyte, mitigating the irreversible lithium loss. Generally, porous copper coatings reduce the electron transfer resistance and exhibit a high diffusion coefficient (10
−6 cm
2 S
−1), thereby improving the rate performance of silicon anodes; however, copper coatings that are excessively thick (>100 nm) can impede the diffusion of Li ions [
76]. Ensafi et al. reported a study on the use of nickel and bismuth as coating materials and highlighted the occurrence of galvanic replacement (which is a type of chemical composition reaction) according to the following half-reaction.
This change was observed only for the bismuth coating, not the nickel coating. The authors suggested that because bismuth is nobler than hydrogen (that is, it has a higher redox potential than that of hydrogen), a successful metal replacement was possible according to the aforementioned reaction; however, in the case of nickel, hydrogen evolution was considerably more dominant than the metal replacement, because of which nickel was coated using a fluoride-containing solution. Similar to that in the Kim et al. study [
74], the silicon that had to be coated was also etched to achieve a large surface area and uniform metal coating. Additionally, the specific capacity, cycling stability, rate performance, and improved CE during cycling were confirmed to improve owing to the enhanced electrical conductivity, similar to that observed for coatings prepared using copper [
71,
74,
75,
76].
Metal oxide coatings (such as Al
2O
3, SiO
2, and TiO
2) are also more promising than metal coatings [
77,
78,
79]. This is because the thin metal oxide coating functions as an artificial SEI layer to suppress the formation of SEI components such as Li
2CO
3, LiF, and other organic components, hence preventing the irreversible consumption of lithium-ions; moreover, it constrains the volume expansion and contraction of silicon particles owing to its increased mechanical integrity [
80]. Jeon et al. reported a study on TiO
2-coated nano-silicon, which was synthesized using the sol–gel method, with an ammonia solution at various pH levels (9.66, 10.14, 10.48,10.70, and 11.08) acting as a catalyst. The amorphous structure of the inactive few-nanometer-thick TiO
2 coating was found to affect electron migration and lithium-ion transfer during the redox reaction between the electrolyte and silicon anode interphase. The coated silicon ranged in size from 60 to 120 nm, and the thickness of the porous TiO
2 coating was determined to be 7–20 nm; upon thermal annealing, the TiO
2 coating did not change in terms of thickness but transformed into an anatase/rutile phase. Furthermore, the pore size and extent of the coating increased with increasing catalyst pH (except at 11.08), and the electron migration was accelerated by the porous coating layer (
Figure 6a). The intrinsic silicon sample exhibited a considerably higher discharge capacity (>2200 mAhg
−1) than that of the other TiO
2-coated silicon anodes in the first cycle (
Figure 6b); this was attributed to the tendency of the TiO
2 coating to inhibit the complete reaction between silicon particles and Li
+ ions. After the second cycle, the TiO
2-coated silicon anode exhibited a significantly higher discharge capacity than that of intrinsic silicon. Among the samples, the TiO
2 coating prepared at a catalyst pH of 10.7 showed optimal cycling performance (
Figure 6b). The coating layer with an appropriately adjusted pore size and dispersion (achieved by tweaking the pH of the catalyst) acted as a buffer to prevent the side reaction between Li ions and silicon and suppressed the volume expansion [
79]. Al
2O
3 has also been extensively investigated as another potent metal oxide coating. For instance, Casino et al. explored the influence of the Al
2O
3 coating on the aging mechanism of silicon anodes (featuring loss of lithium inventory, loss of active material, and increase in cell resistance); this was based on the reported benefits of the Al
2O
3 coating in terms of CE and capacity retention, owing to its tendency to suppress electrolyte decomposition, hence inhibiting the formation of a continuous SEI layer and increasing the mechanical strength. Al
2O
3 coatings with different thicknesses (1.5, 3, and 5 nm) were analyzed for deposition on a 200 nm-thick silicon film. The deposition was conducted by reactive sputtering, and the thickness was adjusted by varying the deposition time. The aging mechanism involved the following events: (1) Loss of lithium inventory occurred owing to SEI formation, irreversible lithium plating, and lithium trapping in the electrically insulated active material, which was related to the irreversible lithium loss. (2) Loss of active material occurred via cracking, metal decomposition, and loss of electrical contact. (3) Incomplete lithiation/delithiation, due to the increase in cell resistance via kinetic limitations. Although the degree to which the Al
2O
3 coating mitigated the loss of the active material was less than that achieved by a carbon coating, it was more effective in reducing lithium-ion consumption owing to the formation of the SEI layer. Therefore, this approach led to a greater decrease in the capacity fading per cycle (64%) than that of the carbon coating (only 29%). Referring to previous research on the aging mechanism of a carbon coating on the silicon anode [
81], the authors emphasized that the Al
2O
3 coating exhibited improved performance in terms of the aging of silicon-based LIB anodes [
78].
SiO
2 also has potential as a metal oxide-type coating material because it can be readily coated and synthesized, in addition to inhibiting the volume expansion because of its mechanical strength and preventing the formation of a continuous SEI layer [
77,
82,
83]. Recently, Schnabel et al. compared a thermally treated SiO
x coating with the native SiO
x wafer layer and found that both specimens exhibited a lower overpotential for lithiation and a thinner, more stable SEI layer than those of the pristine silicon anode. This observation was confirmed from the capacity plot, and decreases in charge consumption were observed for pristine silicon as well as the SiO
x and thermally treated SiO
x layers; however, in the case of native SiO
x, electrolyte decomposition occurred as cycling progressed (
Figure 6c–e). Furthermore, the silicon anodes without the SiO
x coating showed a higher overpotential than that of the specimens with the SiO
x coating (
Figure 6f), indicating that the SiO
x coating inhibited the exposure of silicon to the electrolyte. Additionally, the charge capacity of the native SiO
x coating increased with increasing cycle number, whereas that of the thermal SiO
x-coated silicon decreased; this was because the weaker native SiO
x coating cracked and was then exposed to the electrolyte sooner during cycling (
Figure 6f). Consequently, the coating layer of thermally treated SiO
2 with a thickness of 1–2 nm was deemed optimal for the silicon anode, and the performance of the electrode varied depending on the SiO
2 coating layer [
82].
Nitride-based coating materials have not been studied as much as other inorganic materials such as metals and metal oxides. TiN, a representative nitride-based coating material, is similar to metal, carbon, and conductive polymer layers. In a study conducted on TiN, Tang et al. explored the electrochemical characteristics of TiN-coated silicon synthesized under nitrogen through thermal annealing and its performance compared with TiO
2-coated silicon in a solution. The crystalline TiN coating acted as a conductive layer, provided enhanced conductivity to silicon nanoparticles, and helped form a stable SEI layer, thereby achieving good cycling stability (1900 mAhg
−1 after 100 cycles at 0.1C rate) and rate performance (400 mAhg
−1 at 2C rate). Therefore, the introduction of TiN into the coating material has potential; however, this strategy is not widely used owing to synthesis-related difficulties and the existence of better options [
8].
Overall, metal-type inorganic coatings do not significantly outperform those based on bare silicon. Additionally, because metal coatings are often separated during lithiation/delithiation, they are somewhat expensive owing to the need for pretreatment processes. In the case of metals, improving the conductivity is the predominant aspect that affects the performance enhancement of silicon anodes. Metal oxide- and nitride-type coating materials can outperform metals in terms of improving silicon anodes. Metal oxide coatings act as an artificial SEI layer and are particularly effective in capacity retention and improving the ICE. However, because thickness and porosity affect the degree to which metal oxide coatings contribute to the performance, they should be applied based on experimental data. Furthermore, the mechanical vulnerability of these systems remains unresolved, which could serve as a potential risk factor during long-term cycling.
2.3. Polymer Materials
Polymers have been proposed as alternatives to overcome the disadvantages associated with carbon coatings, which can act as catalysts for electrolyte decomposition due to their high conductivity, and with metal oxide layers, which exhibit weak toughness. Conventional polymers with suitable conductivity and robust properties, such as poly(acrylic acid) (PAA), carboxymethyl cellulose (CMC), and styrene-butadiene rubber (SBR), have been widely used as binders [
84,
85,
86,
87]. These materials improve the electrical contact of silicon nanoparticles through their numerous carboxyl groups, but most polymers are electrical insulators, which can degrade the performance of lithium-ion batteries [
88]. Consequently, conductive polymers are being extensively studied as coating layers. As coating materials, conductive polymers exhibit specific characteristics such as elasticity, toughness, and multifunctional interfaces [
68]. Materials like PEDOT are favored for their ease of processing and high conductivity (1000 S/cm) and have been used to enhance silicon performance either as binders or electrolyte additives. However, instead of these approaches, introducing conductive polymer coatings that can enhance lithium-ion flux for isotropic volume expansion and overcome the low lithium-ion diffusion coefficient has been proposed. Using polymers as coating materials can buffer volume expansion and homogeneously control the uniform interfacial lithium-ion flux in silicon nanoparticles, thus achieving a stable structure. However, the synthesis of polymer organic compounds is complex, necessitating the development of simpler and more cost-effective synthesis methods. Additionally, the wet coating method poses challenges in achieving uniform polymer layers, which can hinder homogeneous lithium-ion movement. In conclusion, the effectiveness of the coating layer heavily depends on the structural and functional characteristics of the coated polymer. The improvements to the silicon anode offered by the coated polymer layer include (a) acting as an artificial SEI layer, (b) enhancing electrolyte wettability, (c) dispersing silicon nanoparticles [
89], (d) improving electrical contact between electrode materials, and (e) controlling the components of the SEI layer [
68,
90,
91,
92,
93,
94].
Recent work by Chen et al. (2023) verified the electrochemical performance of a robust conductive poly(acrylonitrile)-sulfur (PAN-S) layer on a silicon surface, which enables homogeneous Li
+ flux and enhances electrochemical kinetics, depending on the thickness of the coating. The authors emphasized that silicon coated with a flexible PAN-S layer facilitates uniform Li
+ transport by mitigating silicon’s volume expansion and maintaining a stable SEI layer. In contrast, bare silicon showed uneven Li
+ transport, leading to the collapse of silicon particles and the thickening of the SEI layer after cycling. XRD analysis of the PAN-S coating layer revealed its amorphous crystallinity, with sulfur forming short-chain bonds with carbon, similar to organosulfides in PAN-C. Both bare silicon and PAN-S-coated silicon had particle sizes of 50 nm. Three samples with varying PAN-S content were prepared: Si@3%PAN-S with a 3 nm coating layer, Si@6%PAN-S with an uneven 8 nm coating layer, and Si@10%PAN-S with a 13 nm coating layer. The authors highlighted the Li
+ diffusion barrier on silicon surfaces at nitrogen (N) and sulfur (S) sites. Referring to previous data on adsorption energies, they noted higher adsorption at N (−3.51 eV) and S (−3.52 eV) sites compared to Si atoms (−1.03 eV), Si–Si bridges (−1.73 eV), and silicon vacancies (−2.77 eV), leading to more stable Li
+ adsorption at N and S sites. Additionally, the lower Li
+ diffusion barrier indicated easier diffusion at N and S sites, improving rate performance and suggesting that PAN-Li2Si has a high Li
+ transport capability. Consequently, a high-rate capability was confirmed at a 4C-rate current density in PAN-S-coated silicon compared to bare silicon, particularly in Si@6%PAN-S with a capacity of 1103 mAh/g, while bare silicon had only 310 mAh/g at the same rate (
Figure 7c). Furthermore, the two reduction peaks at 1.1 V and 0.75 V observed in bare silicon, indicating electrolyte decomposition (FEC and EC), were less prominent in Si@6%PAN-S, suggesting that the PAN-S coating suppresses electrolyte decomposition. This suppression was attributed to the PAN-S layer preventing direct contact between silicon particles and the electrolyte [
95]. As a result, the initial Coulombic efficiency (ICE) improved from 74% in the bare silicon anode to 81% in the PAN-S-coated silicon anode (
Figure 7a,b). The electrode thickness increased from 12.3 μm to 34.5 μm in bare silicon but showed less improvement in Si@PAN-S electrodes, from 12.8 μm to 25.6 μm, and surface cracks were more severe in bare silicon. The authors concluded that the conductive PAN-S coating layer contributes to the formation of a thin, dense, and robust SEI layer, enhancing Li
+ transport ability and improving the ICE and rate capability of silicon anodes [
68].
In another study by Yoon et al., PAN was used as a coating layer on a silicon anode to assess the wettability of the PAN coating layer in the electrolyte. They confirmed that the PAN coating layer provides ultrafast rate capability and exhibits long-term cycle performance. This is because it prevents localized Li
+ ion insertion and extraction due to the high wettability of the PAN coating layer in a polar solvent. However, a separate structural treatment was required due to the plastic deformation of the PAN coating layer, which led to a reduction in capacity. To address this, nano silicon particles were coated with chemically cross-linkable PAN polymers, as shown in
Figure 7d (SiNP@x-PAN). As a result, homogeneous Li
+ ion transport through wettability, as well as improved cycle capability due to the elastic characteristics, were obtained in SiNP@x-PAN, with only a 23.8% capacity decay after 100 cycles [
94]. This demonstrates that improved properties can be achieved through chemical structure conversion of the polymer layer (e.g., from PAN to cross-linkable PAN copolymer).
Polypyrrole (PPy) is easily synthesized through chemical oxidants such as FeCl3 and has been studied as a coating layer for silicon anodes. It prevents the loss of electrical contact between active materials and current collectors and hinders electrolyte penetration into cracked silicon particles. La et al. prepared PPy-coated silicon through polymerization at room temperature for 24 h after adding poly(N-vinylpyrrolidone), pyrrole, and FeCl3 to etched silicon nanoparticles. The thickness of the PPy coating layer varied depending on the amount of pyrrole, with two comparative groups (2 nm and 4 nm coating layers). Both groups exhibited similar results in CV tests, so the silicon with the 2 nm PPy coating layer was used as the experimental group. In the PPy-coated silicon, a significantly improved discharge capacity was confirmed at each cycle (1000 mAh/g at the 10th cycle, compared to only 130 mAh/g at the 8th cycle for bare silicon). However, the contact between silicon particles and the coating layer became loose within a few cycles, limiting its commercial viability, although the coating was found to prevent cracked silicon particles from being exposed to the electrolyte [
96].
Other studies on silicon anodes coated with polythiophene (PTh), synthesized using an oxidation polymerization method, have demonstrated high capacity and cycle performance. Wang et al. showed that the PTh coating layer, which is a Cα–Cα conjugation-dominant polymer, has good conductivity due to the maximum overlap of the C–C inter-ring carbon pz orbitals. The broad peaks in XRD patterns indicated that the PTh layer was amorphous. When comparing bare silicon and PTh-coated silicon using a Nyquist plot, the semicircle representing charge transfer impedance was smaller for Si@PTh than for bare silicon. Additionally, as the PTh content increased (from 7.5 to 15.2 wt%), the capacity remaining after 50 charge/discharge cycles also increased from 375 mAh/g to 478 mAh/g, as the PTh coating layer improved the electrical contact between silicon particles. However, if the coating was too thick, Li ion insertion and extraction were hindered (when the PTh content increased to 26.2 wt%, the specific capacity of Si@PTh reduced to 224 mAh/g) [
92].
In research using polyurea as a coating material, conducted by Mu et al., it was found that polyurea, with hydrogen bonds and polar functional groups, can be coated through the molecular layer deposition (MLD) method to improve cycle stability, high-rate capability, and Li ion kinetics. Polyurea demonstrates strong adhesion to the silicon surface due to its hydrogen bonds and polar functional groups. If polyurea is repeatedly processed to the desired thickness through MLD instead of directly depositing it, the effect of the deposited layer on electron transport can be increased, as shown in
Figure 7e [
97,
98]. The XPS spectra for bare silicon and polyurea-coated silicon confirmed the N 1 s peak by the C–B bond from the polyurea polymer coating layer. The thickness of the coated layer obtained after 25 cycles (Si@25PU) was 3.0 nm, and the most-improved cycle reversibility (2049 mAh/g at the 80th cycle, with a 69% Coulombic efficiency) was confirmed, making it the experimental group. The Si@25PU electrodes showed improved rate capability (
Figure 7f), with a high reversible capacity of 1820 mAh/g even at 2.0 A/g, due to the effect of the polyurea interface on Li ion diffusion kinetics. Furthermore, the polyurea coating layer has been shown to be applicable to large-sized silicon anodes, helping to improve performance even with ether-based electrolytes (
Figure 7g). The structural stability and integrity of silicon anodes were attributed to the polyurea coating layer, which enhances Li ion kinetics and promotes the formation of a thin, LiF-dominant SEI layer [
90,
99].
2.4. Double Shell
The aforementioned materials have their advantages and disadvantages; however, they exhibit limited performance in enabling the commercialization of silicon anodes with only a single coating, which includes a high percentage of silicon anodes. To overcome this issue, strategies have been devised to prepare coating materials by combining two or more substances, which would solve the problems associated with the various aspects of silicon. This section explores how double-layer coatings prepared from various materials affect silicon anodes and their properties. In particular, the double-layer coatings obtained using two different materials and those prepared by exploiting the differences in the structural and morphological characteristics of the same material have been presented as examples [
100,
101,
102].
For example, a graphitic layer can prevent the exposure of silicon particles to fluoride-anion electrolytes, improve electrical conductivity, and suppress volume expansion; however, a graphitic layer with rigid properties ruptures easily, which is disadvantageous for long-term cycling. To alleviate this issue, Zhu et al. prepared a “soft–hard” double coating whose inner and outer layers comprised a graphitic material as the conductive layer and crosslinked poly(acrylonitrile) (PAN) as an elastic 3D network for relieving the stress experienced by the silicon particles, respectively. The PAN layer had charged nitrile groups and delocalized sp
2 bonding; thus, the coating layer evidently received Li ions and maintained a certain number of electrons and Li ions. The authors prepared Si/C/PAN composites by carbonizing the silicon particles with PAN at 800 ℃ after high-energy ball milling, and the synthesized Si/C (PAN layer converted into conductive carbons) was subsequently cured at 320 ℃ (another coating layer prepared on the Si/C composites). In the case of pristine silicon and Si/C composites, the carboxymethyl cellulose (CMC) binder was weakly bonded to the silicon surface, whereas in the case of Si/C/PAN, the outer PAN layer formed hydrogen bonds with the CMC binder and relieved the stress generated by volume expansion through the 3D networking. Furthermore, the Si/PAN composites showed excellent bonding strength with the binder; however, silicon polarization and SEI formation occurred owing to the absence of a conductive carbon coating. The silicon anode with the double-layer coating exhibited a high initial discharge capacity of 2280 mAhg
−1 at 0.1C and an ICE of 87.38% because the double coating prevented electrolyte decomposition and relieved stress in the electrode. Additionally, a remarkably high CE of 99% was achieved after three cycles, highlighting the long-term cycling characteristics that were due to the robustness of the coating, which was due to the elimination of cracks even after prolonged cycling. Accordingly, the double coating was confirmed to be superior to traditional carbon coatings or polymer coatings [
101].
Lu et al. reported another representative silicon anode that was coated with double layers of carbon. The first carbon coating was formed using a metal–organic framework (MOF), and a graphene network was constructed using silicon nanoparticles encapsulated in ZIF-8; after the synthesis of the first carbon coating and ball milling, a solvothermal reaction and pyrolysis process were conducted (
Figure 8a). The coating layer prepared from ZIF-8 and sucrose exhibited a high ratio of the D and G band intensities, as indicated in the Raman spectrum of graphene/IOC@Si (D band at 1359 cm
−1 and G band at 1591 cm
−1); therefore, the specimen was confirmed to be amorphous, and the outer layer was estimated to be 7 nm thick. The outer carbon coating layer alleviates the volume expansion of silicon nanoparticles, whereas the inner porous carbon layer derived from MOFs with a thickness of ~2 nm provides a favorable pathway for the migration of electrolyte ions, increasing the contact between the internal silicon nanoparticles and electrolyte ions, enabling improvements in capacity and rate performance (
Figure 8b). Additionally, an analysis of the lithium storage mechanism and diffusion dynamics confirmed that the designed double-layer carbon coating—particularly the inner carbon layer—effectively promoted the diffusion of Li ions to the silicon surface, leading to reduced resistance in the Nyquist plot (
Figure 8c) [
100]. Another study on a double-layer coating, which contained thin SiO
2 and carbon on silicon nanoparticles, was conducted by Zhang et al. The conventional carbon-coated silicon anode did not have sufficient mechanical strength to accommodate the force generated by the volume expansion during charging/discharging. To synthesize Si@SiO
2@C with a core/double-shell structure featuring an interlayer of SiO
2 and an outer layer of C (
Figure 8d), thin SiO
2 and C layers were coated on the surface of silicon nanoparticles via the hydrolysis of tetraethyl orthosilicate combined with the carbonization of polydopamine. As shown in
Figure 8d, both Si@C and Si@SiO
2@C showed greater cycling stability with higher capacity (401 and 798 mAhg
−1, respectively, after 50 cycles) than those of pristine silicon (only 303 Ahg
−1 after 50 cycles). However, among the two coated samples, Si@SiO
2@C exhibited greater capacity retention than that of Si@C. This observation was attributed to the mechanical strength of pristine silicon, and Si@C could not sufficiently withstand the force generated during lithiation and delithiation, leading to a crack on the surface of the silicon anode after 50 cycles at 0.1 Ag
−1. By contrast, the SiO
2 interlayer in Si@SiO
2@C acted as a mechanical clamping layer to limit the volume change of the silicon core during lithiation and delithiation, thereby ensuring remarkably stable cycling performance; even the inner layer consumed lithium, forming irreversible Li
4SiO
4. An analysis of rate capability (
Figure 8f) indicated that the Si@SiO
2@C electrode exhibited relatively stable capacities (870.4, 856.2, 757.1, 650.7, 528.3, and 353.4 mAhg
−1 at current densities of 0.1, 0.2, 0.5, 1.0 2.0, and 5.0 Ag
−1, respectively) than those of Si@C (376.8, 328.2, 249.4, 189.8, 135.5, and 32.8 mAhg
−1). The improved rate capability was achieved because the additional SiO
2 coating helped increase the graphite content and electrical conductivity, leading to a reduced
Rct. Consequently, the double coating prepared using SiO
2 and carbon enabled the formation of a stable SEI layer by increasing the mechanical strength, helped maintain adequate electrical contact with the electrode, and formed a dense carbon layer, thereby increasing electrical conductivity [
103]. These results indicated that complementary coatings can be realized by achieving more performance improvements with different constituent materials.
The double-layer coating is significant, as it is constructed by leveraging the complementary attributes of two coating layers exhibiting varying performance effects. Accordingly, several studies are being performed on double-layer coating to achieve good performance [
102,
103,
104]. However, as the coating is double-layered, experimental confirmation of the performance improvement is essential, and the complexity of the process should be reduced.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}