

Hydrogen-Free Deoxygenation of Oleic Acid and Industrial Vegetable Oil Waste on CuNiAl Catalysts for Biofuel Production

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalyst Preparation
2.3. Characterization
2.4. Fast Pyrolysis Experiments
3. Results
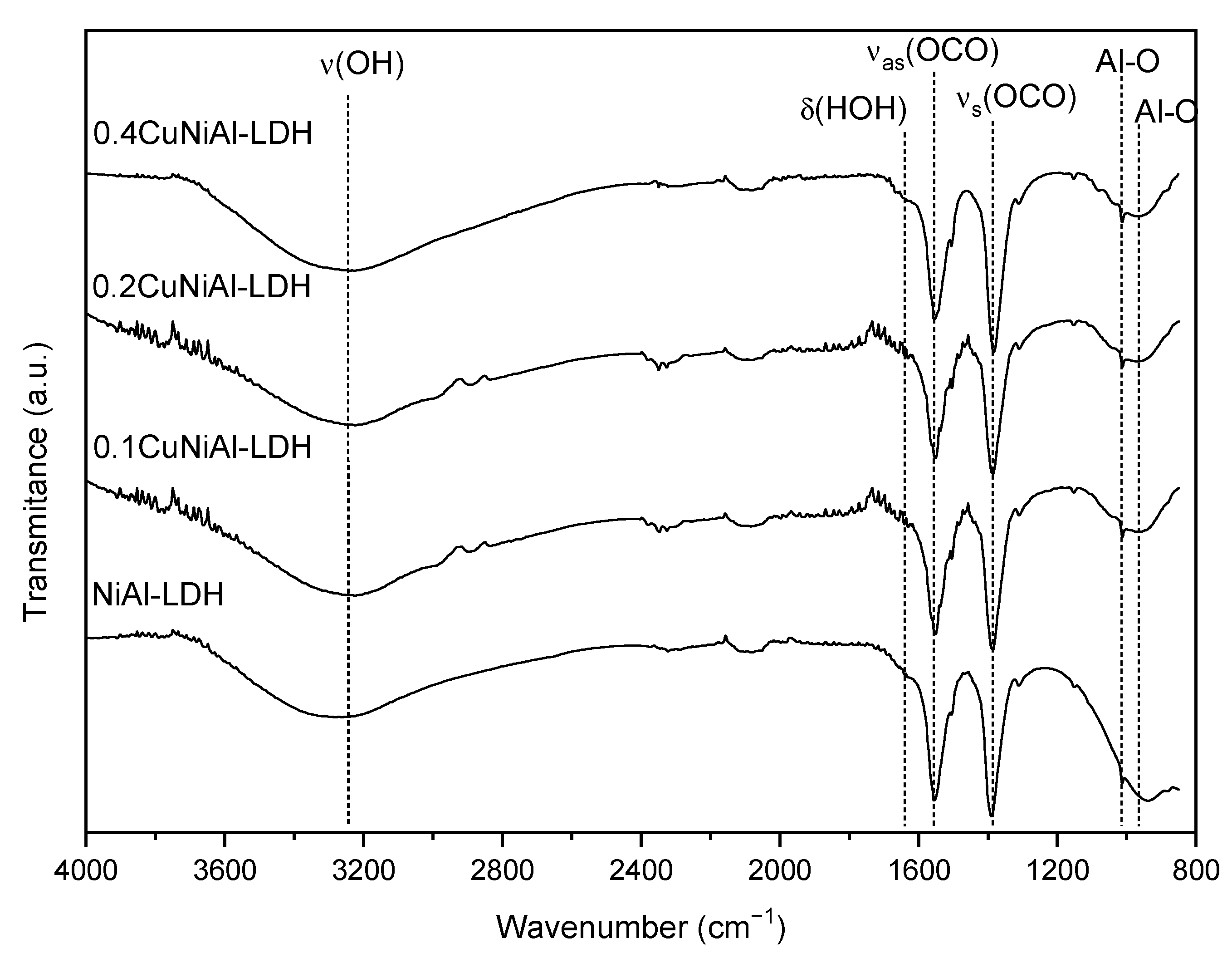
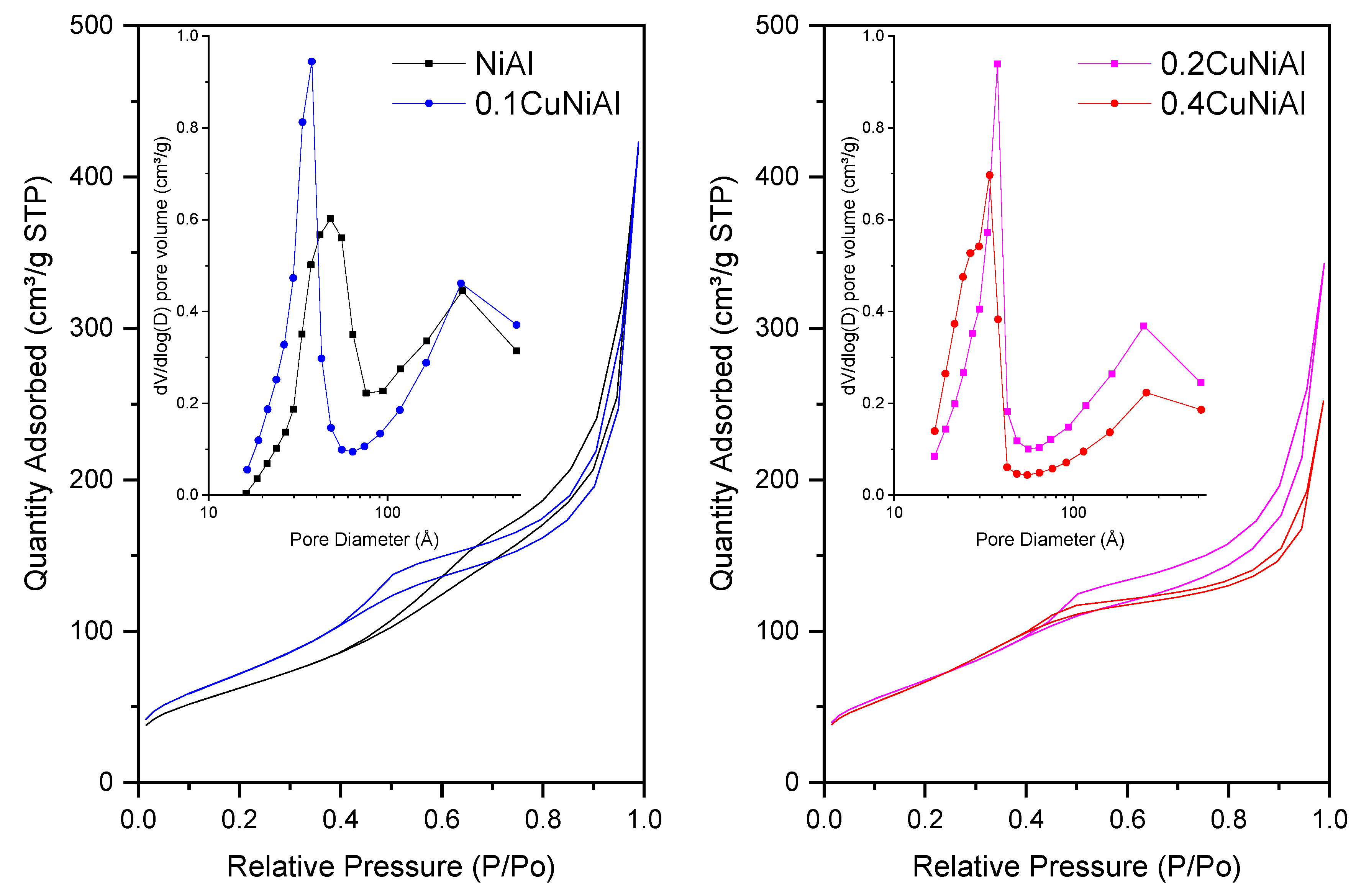
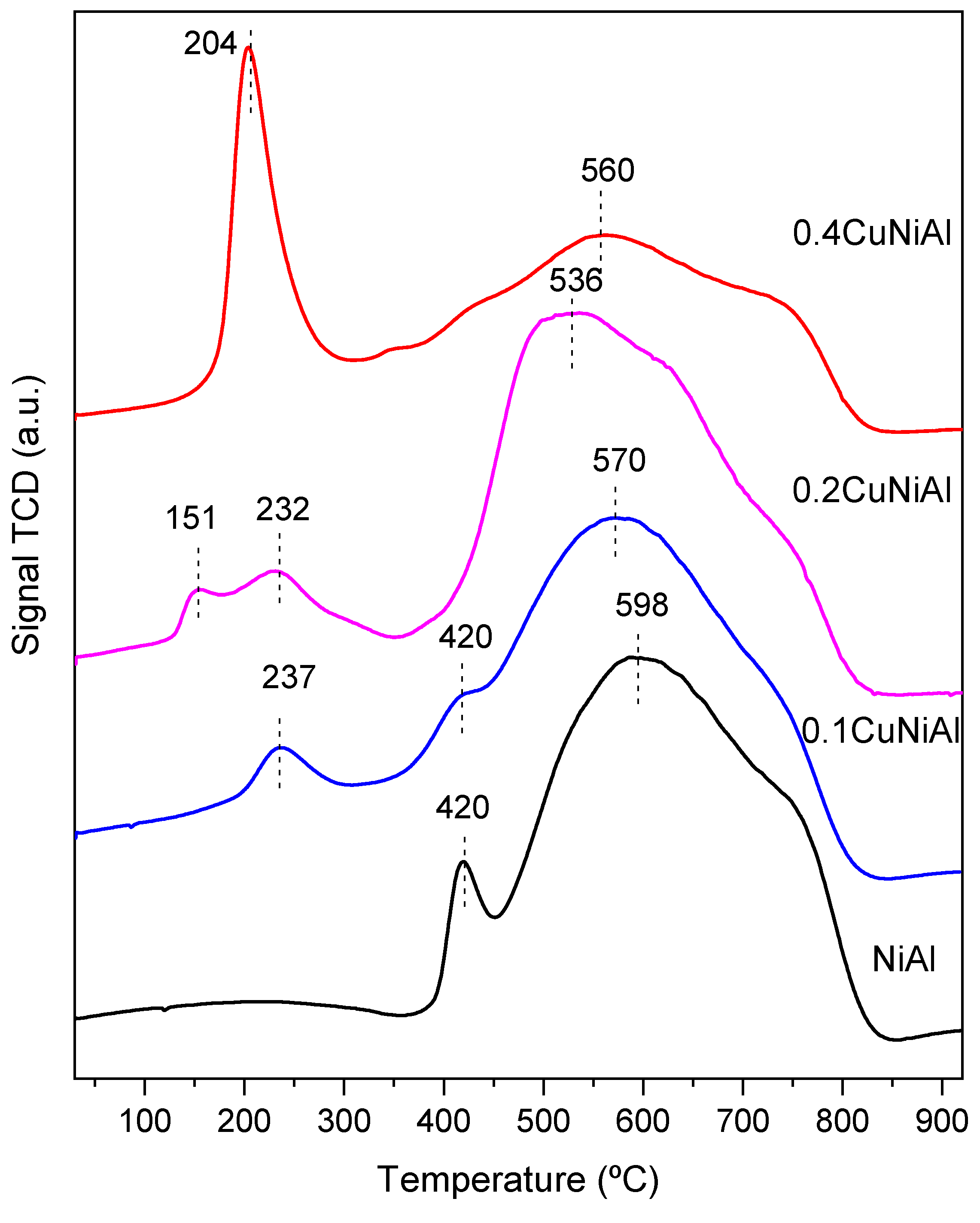
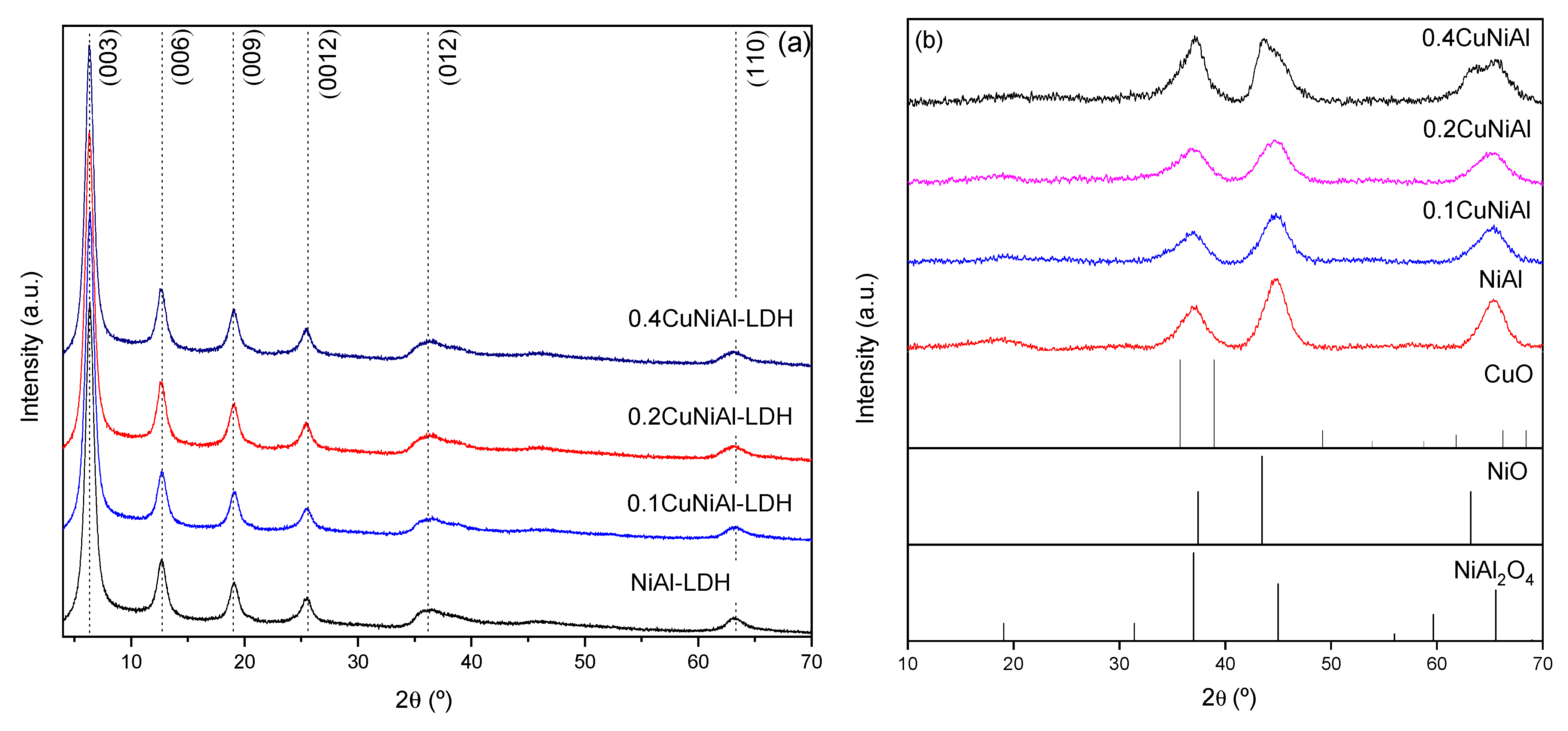
3.1. Characterization of Precursors and Catalysts
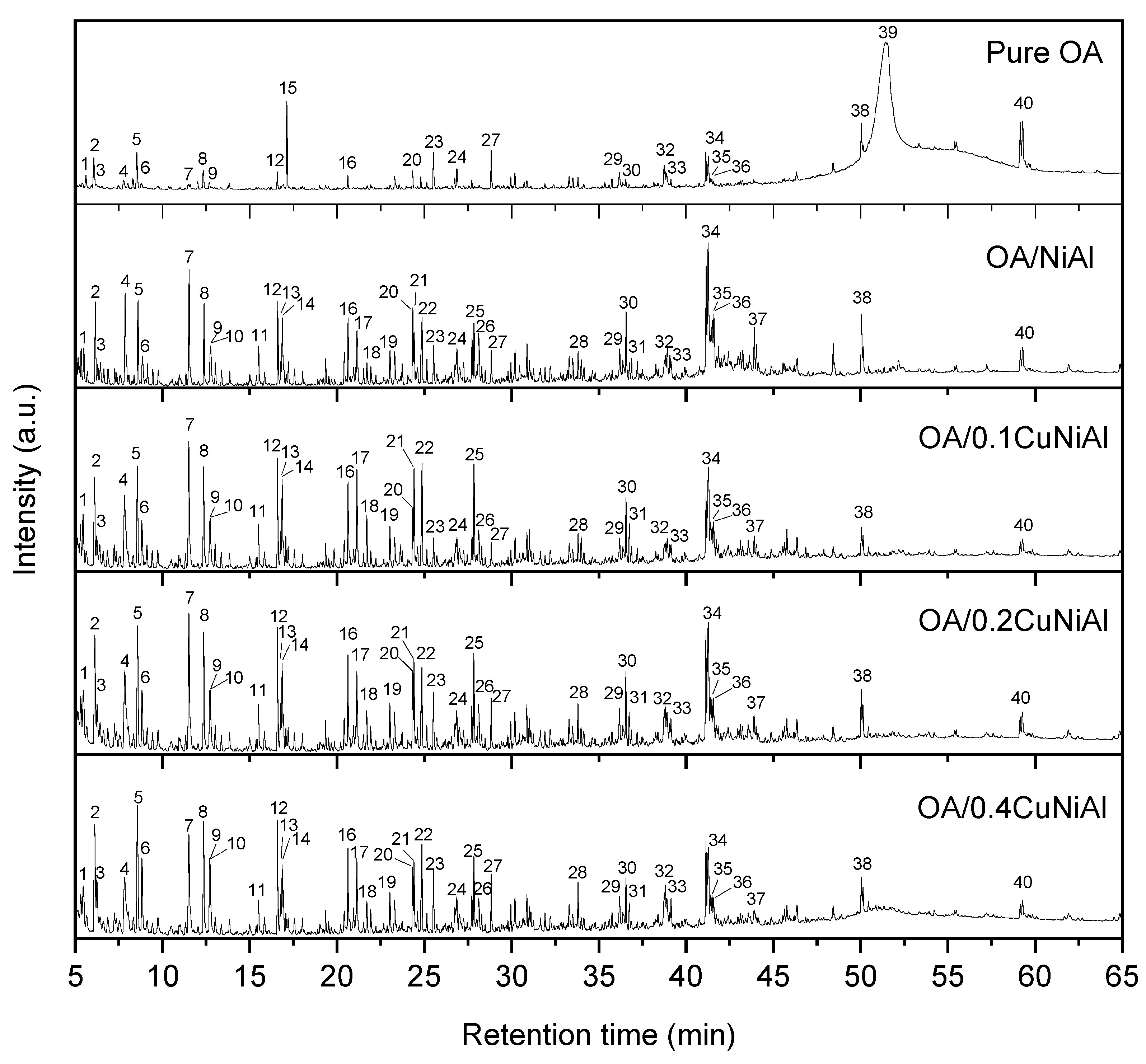
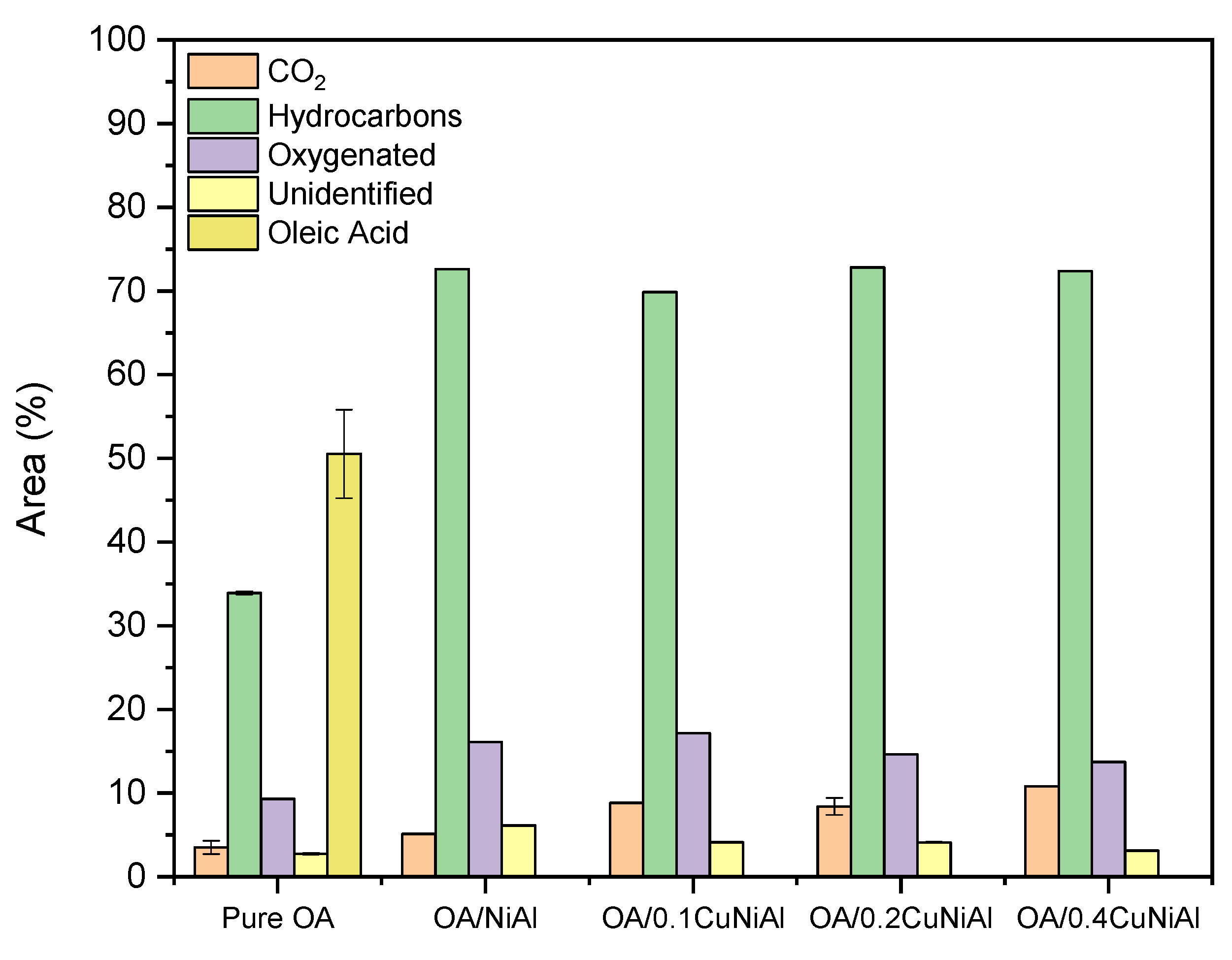
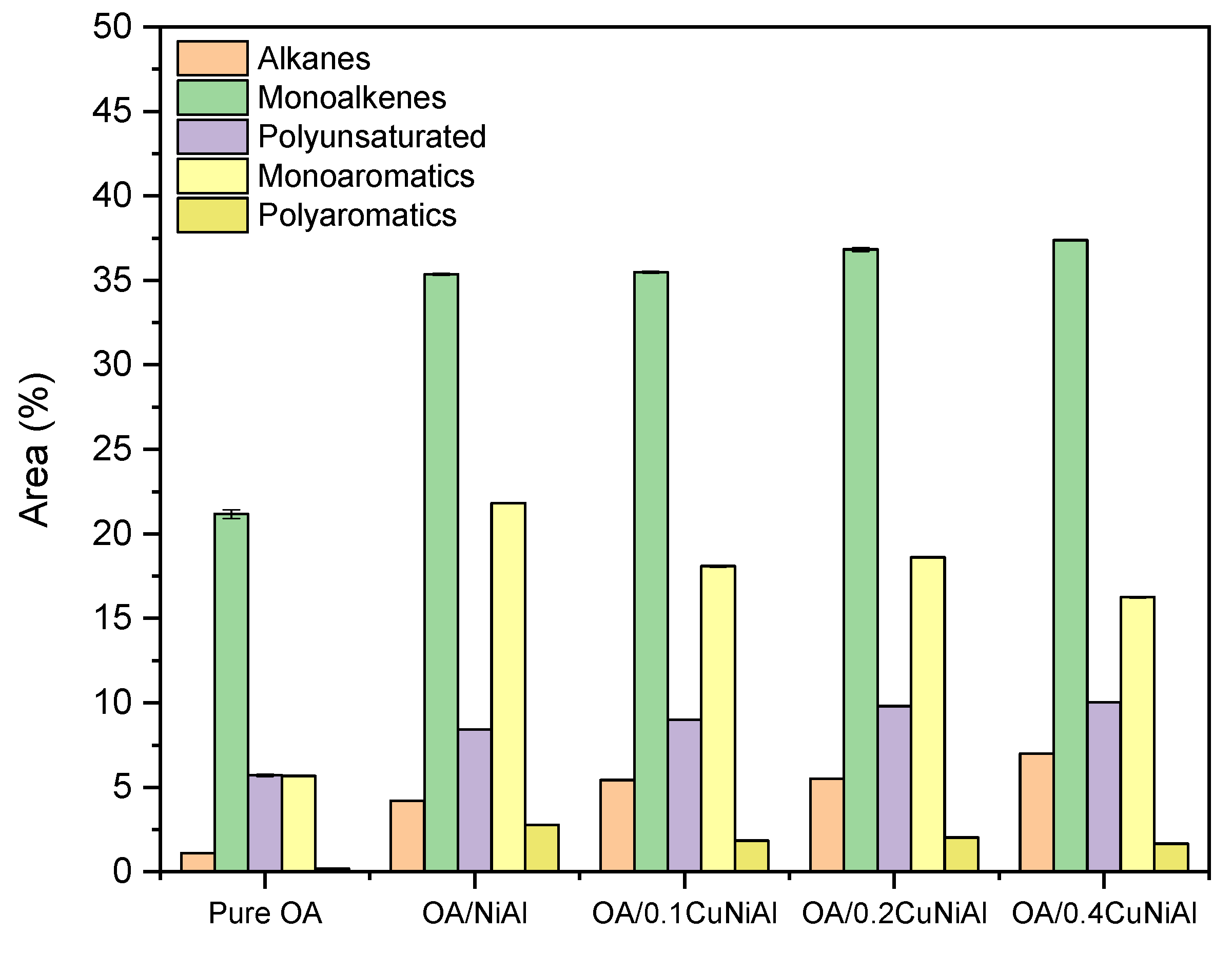
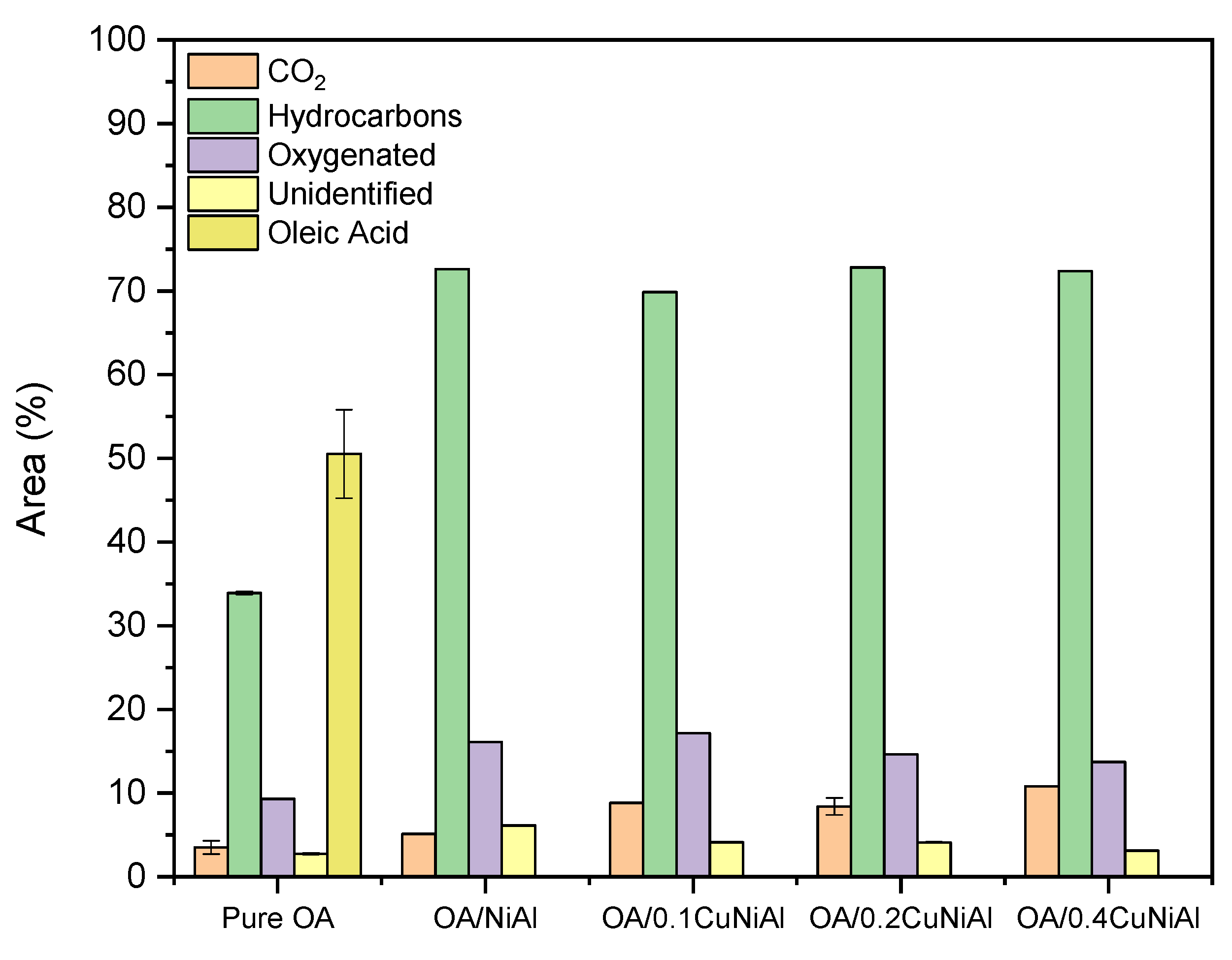
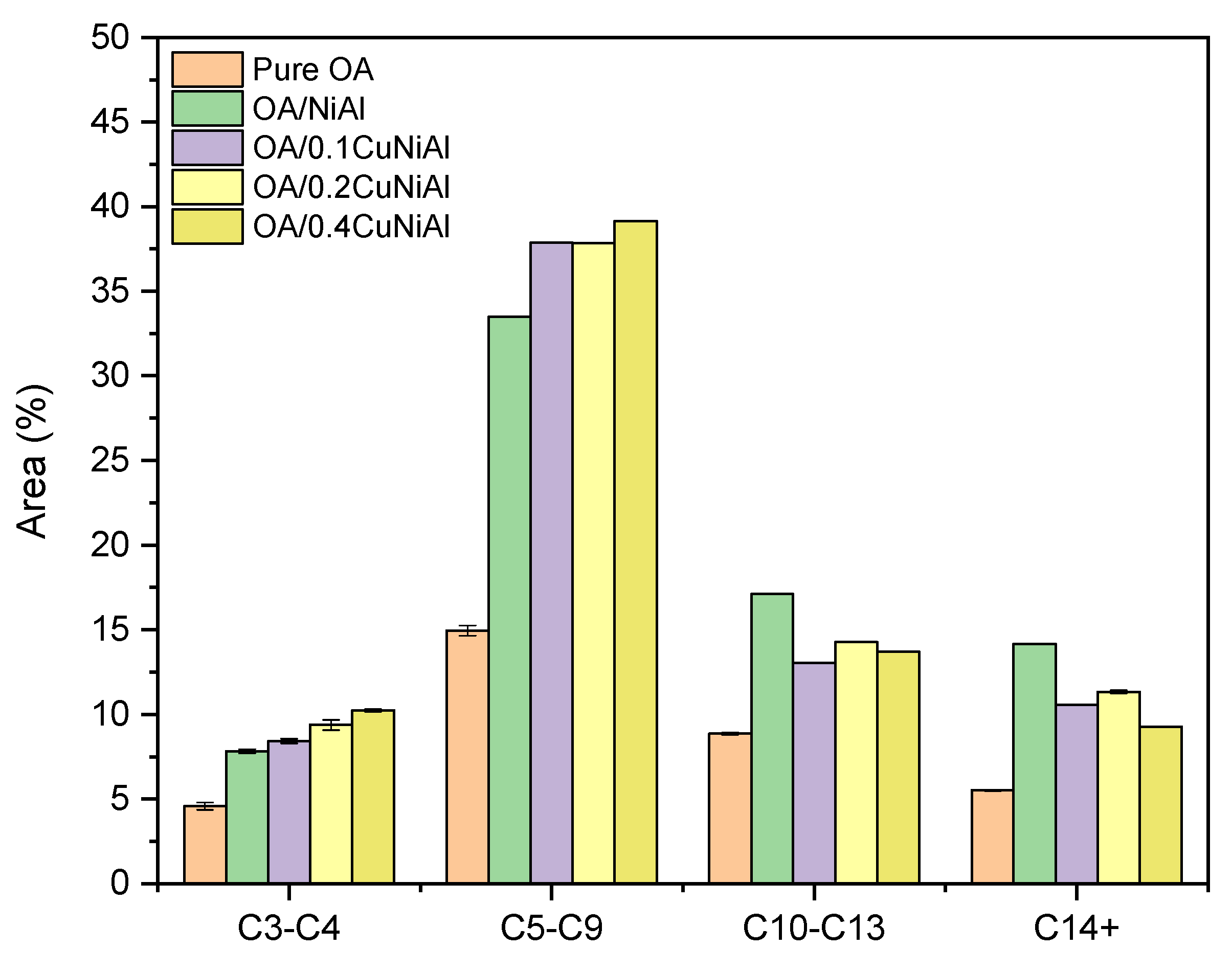
3.2. Oleic Acid Pyrolysis
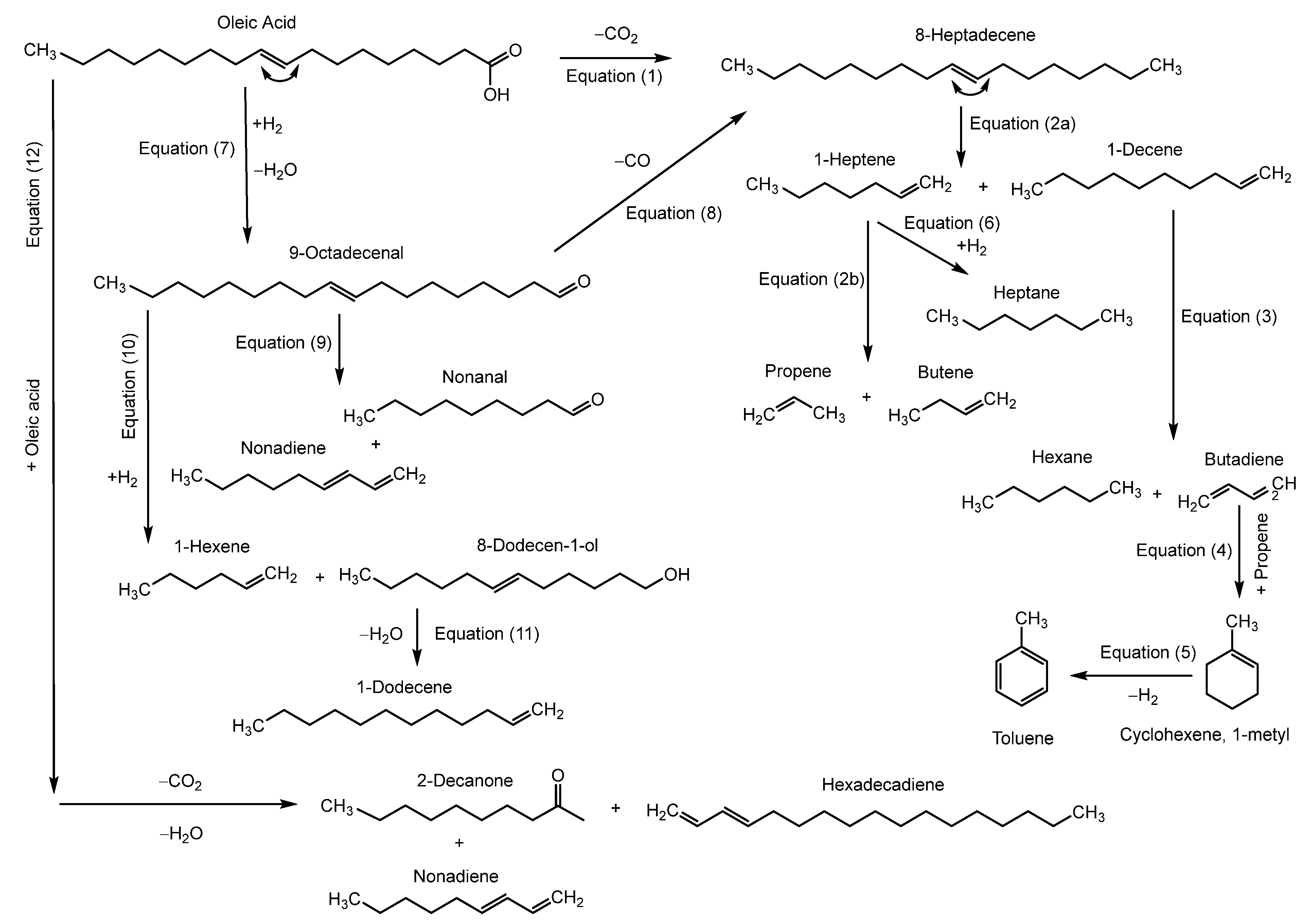
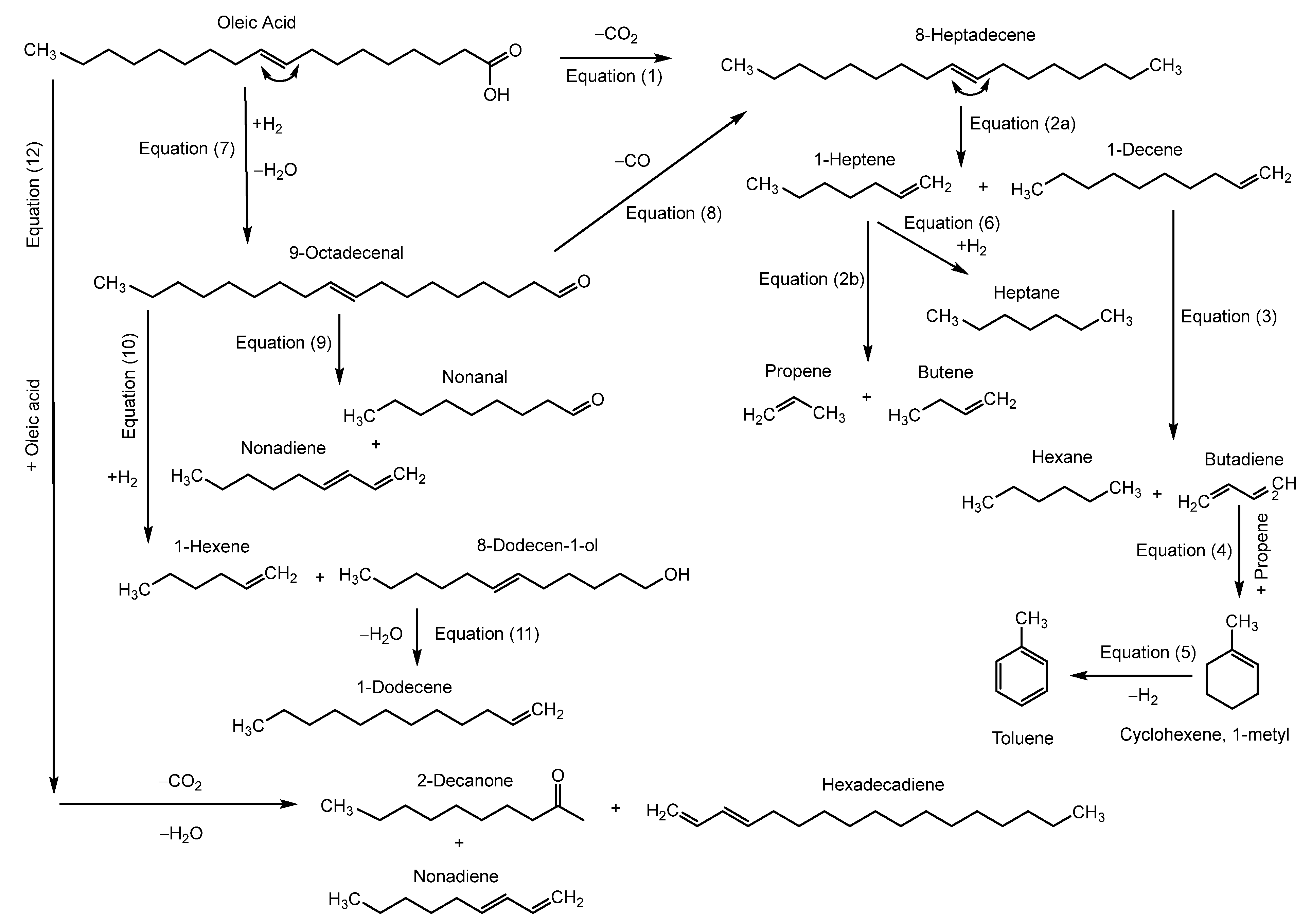
3.3. Reaction Pathways
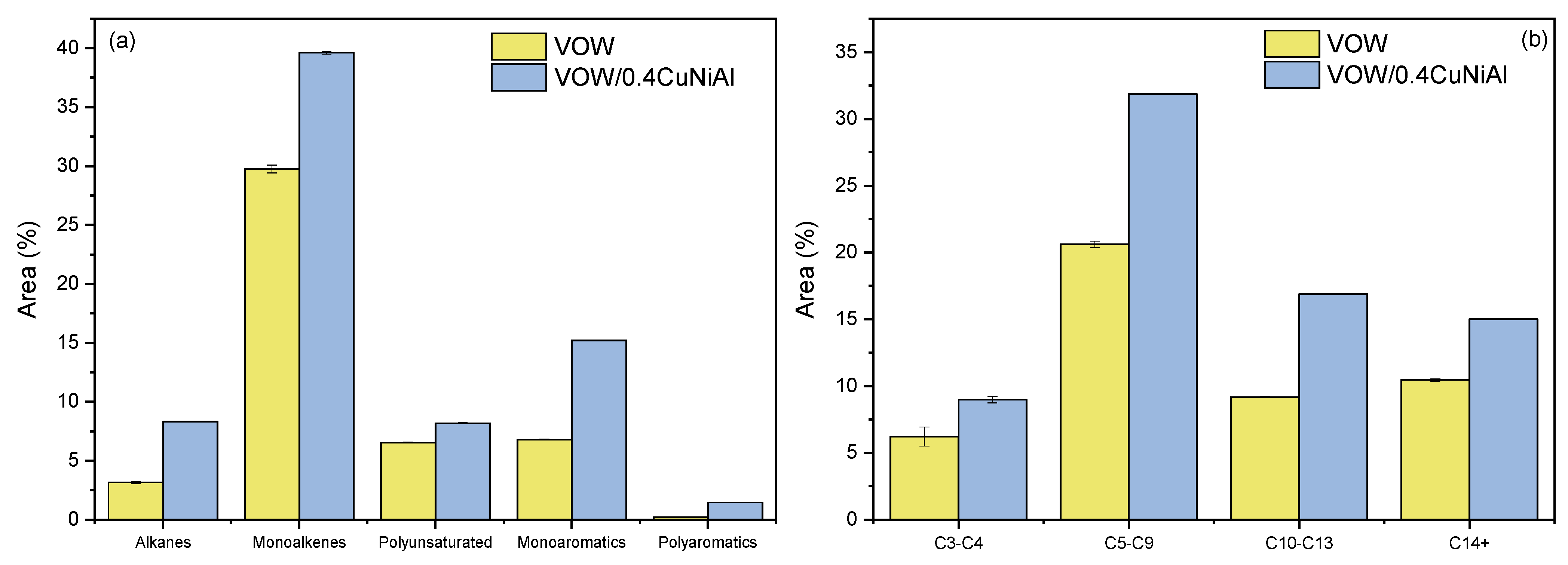
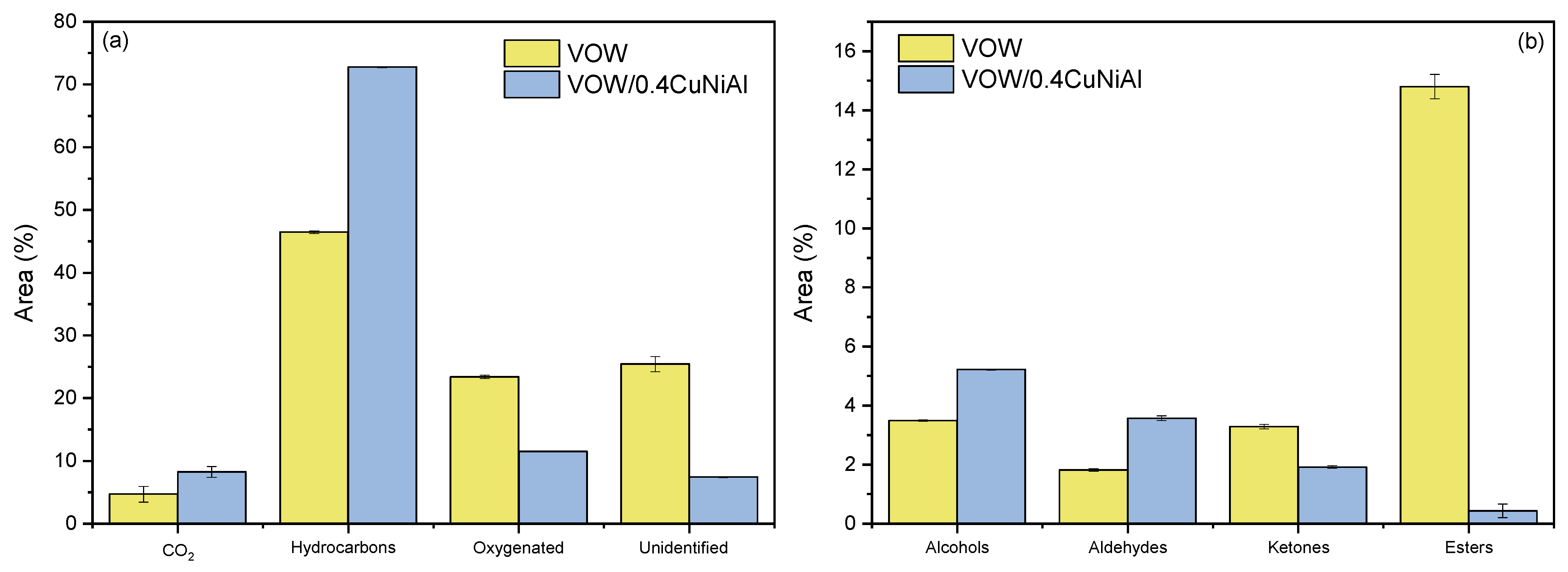
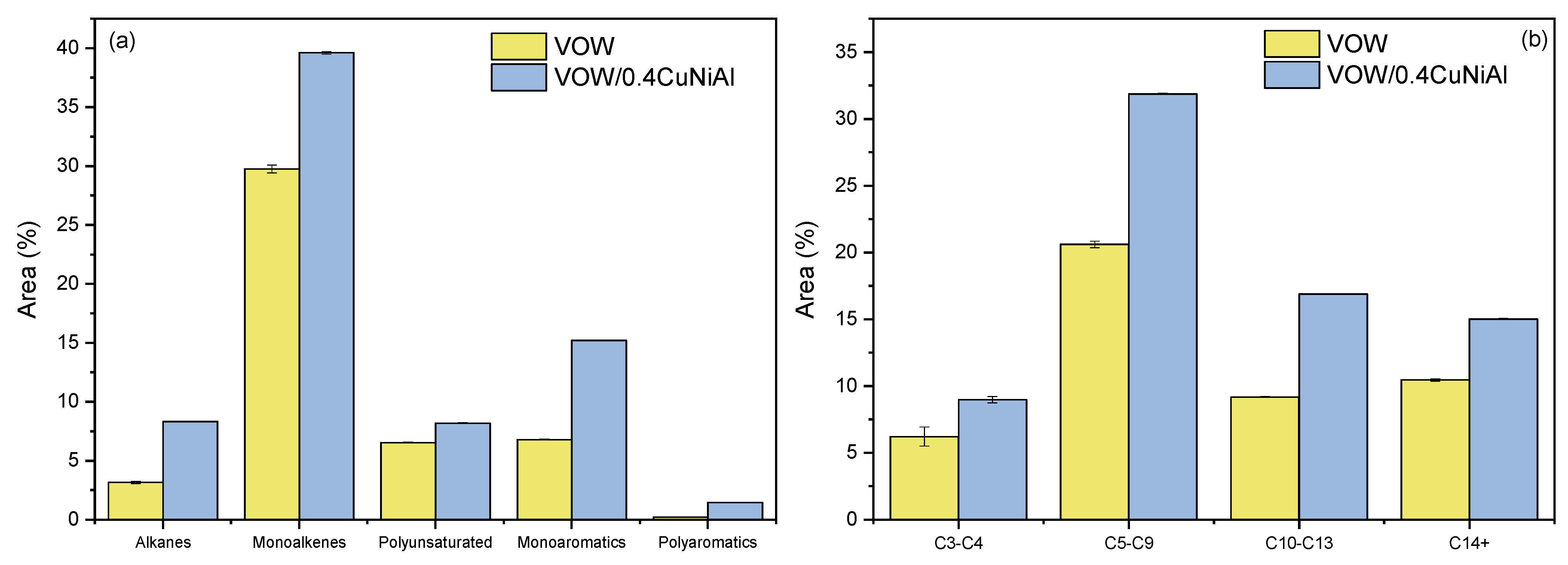
3.4. Vegetable Oil Waste Pyrolysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lucantonio, S.; Di Giuliano, A.; Rossi, L.; Gallucci, K. Green Diesel Production via Deoxygenation Process: A Review. Energies 2023, 16, 844. [Google Scholar] [CrossRef]
- Silva, R.J.M.C.L.; Souza, T.P.C.; Elihimas, D.R.M.; Silva, J.P.; Albuquerque, A.A.; Pacheco, J.G.A.; Silva, J.M.F. Ethanol Dehydration by Absorption and Biodiesel Production by Reactive Distillation: An Innovative Integrated Process. Biomass Bioenergy 2021, 154, 106263. [Google Scholar] [CrossRef]
- Souza, T.P.C.; Silva, R.J.M.C.L.; Melo, J.C.; Tschoeke, I.C.P.; Silva, J.P.; Pacheco, J.G.A.; Silva, J.M.F. Kinetic Modeling of Cottonseed Oil Transesterification with Ethanol. React. Kinet. Mech. Catal. 2019, 128, 707–722. [Google Scholar] [CrossRef]
- Eremeeva, A.M.; Kondrasheva, N.K.; Khasanov, A.F.; Oleynik, I.L. Environmentally Friendly Diesel Fuel Obtained from Vegetable Raw Materials and Hydrocarbon Crude. Energies 2023, 16, 2121. [Google Scholar] [CrossRef]
- Kondrasheva, N.K.; Eremeeva, A. Production of Biodiesel Fuel from Vegetable Raw Materials. J. Min. Inst. 2023, 260, 248–256. [Google Scholar] [CrossRef]
- Farias, A.F.F.; Moura, K.F.; Souza, J.K.; Lima, R.O.; Nascimento, J.D.; Cutrim, A.A.; Longo, E.; Araujo, A.S.; Carvalho-Filho, J.R.; Souza, A.G.; et al. Biodiesel obtained by ethylic transesterification using CuO, ZnO and CeO2 supported on bentonite. Fuel 2015, 160, 357–365. [Google Scholar] [CrossRef]
- de Almeida, R.P.; Aciole, R.C.G.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Pacheco, J.G.A.; Barros, I.C.L. Residue-Based Activated Carbon from Passion Fruit Seed as Support to H3PW12O40 for the Esterification of Oleic Acid. J. Clean. Prod. 2021, 282, 124477. [Google Scholar] [CrossRef]
- de Oliveira, A.D.N.; Ferreira, I.M.; Jimenez, D.E.Q.; da Silva, L.S.; da Costa, A.A.F.; Pires, L.H.D.O.; Luque, R.; Osman, S.M.; Da Costa, C.E.F.; Rocha Filho, G.N.; et al. Esterification of an Agro-Industrial Waste on Kaolinite-Derived Catalyst Prepared via Microwave Irradiation. Waste Biomass Valori. 2022, 13, 3933–3944. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Q.; Ke, L.; Peng, Y.; Liu, Y.; Wu, Q.; Tian, X.; Dai, L.; Ruan, R.; Jiang, L. Review on the Catalytic Pyrolysis of Waste Oil for the Production of Renewable Hydrocarbon Fuels. Fuel 2021, 283, 119170. [Google Scholar] [CrossRef]
- Kurowska, K.; Marks-Bielska, R.; Bielski, S.; Kryszk, H.; Jasinskas, A. Food Security in the Context of Liquid Biofuels Production. Energies 2020, 13, 6247. [Google Scholar] [CrossRef]
- Kumar, A.; Anushree; Kumar, J.; Bhaskar, T. Utilization of Lignin: A Sustainable and Eco-Friendly Approach. J. Energy Inst. 2020, 93, 235–271. [Google Scholar] [CrossRef]
- Ong, H.C.; Chen, W.H.; Singh, Y.; Gan, Y.Y.; Chen, C.Y.; Show, P.L. A State-of-the-Art Review on Thermochemical Conversion of Biomass for Biofuel Production: A TG-FTIR Approach. Energy Convers. Manag. 2020, 209, 112634. [Google Scholar] [CrossRef]
- Amaral, A.R.; Bernar, L.P.; Ferreira, C.C.; de Oliveira, R.M.; Pereira, A.M.; Pereira, L.M.; Santos, M.C.; Assunção, F.P.d.C.; Bezerra, K.C.A.; Almeida, H.S.; et al. Economic Feasibility Assessment of the Thermal Catalytic Process of Wastes: Açaí Seeds (Euterpe Oleracea) and Scum from Grease Traps. Energies 2022, 15, 7718. [Google Scholar] [CrossRef]
- Melo, J.A.; de Sá, M.S.; Moral, A.; Bimbela, F.; Gandía, L.M.; Wisniewski, A. Renewable Hydrocarbon Production from Waste Cottonseed Oil Pyrolysis and Catalytic Upgrading of Vapors with Mo-Co and Mo-Ni Catalysts Supported on γ-Al2O3. Nanomaterials 2021, 11, 1659. [Google Scholar] [CrossRef] [PubMed]
- Azahar, A.A.; Nurhafizah, M.D.; Abdullah, N.; Ul-Hamid, A. A Review on the Palm Oil Waste Thermal Degradation Analysis and Its Kinetic Triplet Study. Bioenergy Res. 2023. [Google Scholar] [CrossRef]
- Su, G.; Mohd Zulkifli, N.W.; Ong, H.C.; Ibrahim, S.; Bu, Q.; Zhu, R. Pyrolysis of Oil Palm Wastes for Bioenergy in Malaysia: A Review. Renew. Sust. Energ. Rev. 2022, 164, 112554. [Google Scholar] [CrossRef]
- Yu, M.; Zhang, C.; Li, X.; Liu, Y.; Li, X.; Qu, J.; Dai, J.; Zhou, C.; Yuan, Y.; Jin, Y.; et al. Pyrolysis of Vegetable Oil Soapstock in Fluidized Bed: Characteristics of Thermal Decomposition and Analysis of Pyrolysis Products. Sci. Total Environ. 2022, 838, 155412. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.R.; Bernar, L.P.; Ferreira, C.C.; Pereira, A.M.; Dos Santos, W.G.; Pereira, L.M.; Santos, M.C.; Assunção, F.P.d.C.; Mendonça, N.M.; Pereira, J.A.R.; et al. Economic Analysis of Thermal–Catalytic Process of Palm Oil (Elaeis Guineesensis, Jacq) and Soap Phase Residue from Neutralization Process of Palm Oil (Elaeis Guineensis, Jacq). Energies 2023, 16, 492. [Google Scholar] [CrossRef]
- da Silva Almeida, H.; Correa, O.A.; Ferreira, C.C.; Ribeiro, H.J.; de Castro, D.A.R.; Pereira, M.S.; Mâncio, A.A.; Santos, M.C.; da Mota, S.A.P.; da Silva Souza, J.A.; et al. Diesel-like hydrocarbon fuels by catalytic cracking of fat, oils, and grease (FOG) from grease traps. J. Energy Inst. 2017, 90, 337–354. [Google Scholar] [CrossRef]
- Andrade, L.A.; Barrozo, M.A.S.; Vieira, L.G.M. Thermo-chemical behavior and product formation during pyrolysis of mango seed shell. Ind. Crops Prod. 2016, 85, 174–180. [Google Scholar] [CrossRef]
- Barbosa, J.M.; Rossi, R.A.S.; Andrade, L.A.; Barrozo, M.A.S.; Vieira, L.G.M. A study of optimization of solar pyrolysis and catalyst recovery and reuse. Energy Conv. Managem. 2021, 237, 114094. [Google Scholar] [CrossRef]
- Bernar, L.P.; Ferreira, C.C.; Costa, A.F.; Ribeiro, H.J.; dos Santos, W.G.; Pereira, L.M.; Pereira, A.M.; Moraes, N.L.; Assunção, F.P.; Mota, S.A.; et al. Catalytic Upgrading of Residual Fat Pyrolysis Vapors over Activated Carbon Pellets into Hydrocarbons-like Fuels in a Two-Stage Reactor: Analysis of Hydrocarbons Composition and Physical-Chemistry Properties. Energies 2022, 15, 4587. [Google Scholar] [CrossRef]
- Wisniewski, A.; Wosniak, L.; Scharf, D.R.; Wiggers, V.R.; Meier, H.F.; Simionatto, E.L. Upgrade of Biofuels Obtained from Waste Fish Oil Pyrolysis by Reactive Distillation. J. Braz. Chem. Soc. 2015, 26, 224–232. [Google Scholar] [CrossRef]
- Kay Lup, A.N.; Abnisa, F.; Wan Daud, W.M.A.; Aroua, M.K. A Review on Reactivity and Stability of Heterogeneous Metal Catalysts for Deoxygenation of Bio-Oil Model Compounds. J. Ind. Eng. Chem. 2017, 56, 1–34. [Google Scholar] [CrossRef]
- Su, G.; Ong, H.C.; Fattah, I.M.R.; Ok, Y.S.; Jang, J.H.; Wang, C.T. State-of-the-Art of the Pyrolysis and Co-Pyrolysis of Food Waste: Progress and Challenges. Sci. Total Environ. 2022, 809, 151170. [Google Scholar] [CrossRef]
- Santos, T.M.; Silva, W.R.D.; Carregosa, J.D.C.; Schmitt, C.C.; Moreira, R.; Raffelt, K.; Dahmen, N.; Wisniewski, A., Jr. Thermal Conversion of Sugarcane Bagasse Coupled with Vapor Phase Hydrotreatment over Nickel-Based Catalysts: A Comprehensive Characterization of Upgraded Products. Catalysts 2022, 12, 355. [Google Scholar] [CrossRef]
- Silva, N.K.G.; Ferreira, R.A.; Ribas, R.M.; Monteiro, R.S.; Barrozo, M.A.S.; Soares, R.R. Gas-phase hydrodeoxygenation (HDO) of guaiacol over Pt/Al2O3 catalyst promoted by Nb2O5. Fuel 2021, 287, 119509. [Google Scholar] [CrossRef]
- Hasanudin, H.; Asri, W.R.; Fanani, Z.; Adisti, S.J.; Hadiah, F.; Maryana, R.; Al Muttaqii, M.; Zhu, Z.; Machado, N.T. Facile Fabrication of SiO2/Zr Assisted with EDTA Complexed-Impregnation and Templated Methods for Crude Palm Oil to Biofuels Conversion via Catalytic Hydrocracking. Catalysts 2022, 12, 1522. [Google Scholar] [CrossRef]
- do Nascimento, L.A.; Barroso-Martín, L.; Peçanha, S.R.S.; Arias, S.; Santos, B.S.; Pacheco, J.G.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Barros, I.C.L. NiAlCe Mixed Oxides Obtained from Layered Double Hydroxides Applied to Anisole Hydrodeoxygenation. Catal. Today 2022, 394–396, 282–294. [Google Scholar] [CrossRef]
- Su, G.; Ong, H.C.; Mofijur, M.; Mahlia, T.M.I.; Ok, Y.S. Pyrolysis of Waste Oils for the Production of Biofuels: A Critical Review. J. Hazard Mater. 2022, 424, 127396. [Google Scholar] [CrossRef]
- Dada, T.K.; Sheehan, M.; Murugavelh, S.; Antunes, E. A Review on Catalytic Pyrolysis for High-Quality Bio-Oil Production from Biomass. Biomass Conv. Bioref. 2023, 13, 2595–2614. [Google Scholar] [CrossRef]
- da Silveira Rossi, R.A.; Barbosa, J.M.; Barrozo, M.A.S.; Vieira, L.G.M. Catalytic solar hydropyrolysis of the Chlamydomonas reinhardtii microalgae. Biomass Bioenergy 2021, 152, 106183. [Google Scholar] [CrossRef]
- Wang, Y.; Ke, L.; Peng, Y.; Yang, Q.; Du, Z.; Dai, L.; Zhou, N.; Liu, Y.; Fu, G.; Ruan, R.; et al. Characteristics of the Catalytic Fast Pyrolysis of Vegetable Oil Soapstock for Hydrocarbon-Rich Fuel. Energy Convers. Manag. 2020, 213, 112860. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Jin, X.; Wang, F.; Shen, B. Synthetic Strategies and Performance of Catalysts for Pyrolytic Production of Alternative Aviation Fuels Using Non-Edible Lipids: A Critical Review. Appl. Catal. A Gen. 2022, 643, 118769. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Fonseca, F.G.; Fraga, M.M.C.; Wisniewski, A., Jr.; Karp, S.; Mello José, A.H.; Rodrigues, R.C.L.B.; Moreira, R.; Hirayama, D.E.; Raffelt, K.; et al. Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization. Catalysts 2021, 11, 1549. [Google Scholar] [CrossRef]
- Silva, L.D.; Lira, T.S.; Xavier, T.P.; Barrozo, M.A.S.; Dantas, S.C.; Silvério, B.C.; Santos, K.G. Effect of Temperature and MgCl2 Concentration on the Catalytic Pyrolysis of Malt Waste Using Response Surface Methodology. ChemistrySelect 2022, 7, e202200663. [Google Scholar] [CrossRef]
- Silva, F.C.M.; Lima, M.S.; Costa Neto, C.O.; Sá, J.L.S.; Souza, L.D.; Caldeira, V.P.S.; Santos, A.G.D.; Luz, G.E., Jr. Catalytic deoxygenation of C18 fatty acids over HAlMCM-41 molecular sieve. Biomass Convers. Biorefin. 2018, 8, 159–167. [Google Scholar] [CrossRef]
- Arias, S.; Vascocelos, D.P.; Libório, D.O.; Gonzalez, J.F.; Câmara, A.G.; Barbosa, C.M.B.M.; Fréty, R.; Pacheco, J.G.A. Hydrogen-Free Deoxygenation of Industrial Vegetable Oil Waste Using Ce, Zr-NiAl Catalysts for Second-Generation Biofuels Production. Mol. Catal. 2022, 529, 112554. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, J.; Liu, C.; Lu, Y.; Lin, X.; Li, W.; Zheng, Z. Efficient and Stable Ni-Cu Catalysts for Ex Situ Catalytic Pyrolysis Vapor Upgrading of Oleic Acid into Hydrocarbon: Effect of Catalyst Support, Process Parameters and Ni-to-Cu Mixed Ratio. Renew. Energy 2020, 154, 797–812. [Google Scholar] [CrossRef]
- Shafizadeh, A.; Rastegari, H.; Shahbeik, H.; Mobli, H.; Pan, J.; Peng, W.; Li, G.; Tabatabaei, M.; Aghbashlo, M. A Critical Review of the Use of Nanomaterials in the Biomass Pyrolysis Process. J. Clean. Prod. 2023, 400, 136705. [Google Scholar] [CrossRef]
- Khalid, M.A.A.; Abdullah, N.; Ibrahim, M.N.M.; Taib, R.M.; Rosid, S.J.M.; Shukri, N.M.; Yahaya, N.F.; Abdullah, W.N.B.W. Catalytic Pyrolysis of Waste Oil into Hydrocarbon Fuel Utilizing Cerium Oxide Catalyst. Korean J. Chem. Eng. 2022, 39, 1487–1495. [Google Scholar] [CrossRef]
- Nabgan, W.; Ikram, M.; Alhassan, M.; Owgi, A.H.K.; Van Tran, T.; Parashuram, L.; Nordin, A.H.; Djellabi, R.; Jalil, A.A.; Medina, F.; et al. Bibliometric Analysis and an Overview of the Application of the Non-Precious Materials for Pyrolysis Reaction of Plastic Waste. Arab. J. Chem. 2023, 16, 104717. [Google Scholar] [CrossRef]
- Çakman, G.; Ceylan, S.; Balci, S. Catalytic Deoxygenation of Oleic Acid over Synthesized Ni@CMK-3 Catalyst Using Analytical Py-GC/MS and TG-FTIR. J. Porous Mater. 2023, 30, 899–909. [Google Scholar] [CrossRef]
- Arias, S.; González, J.F.; Sousa, L.V.; Barbosa, C.B.M.; Silva, A.O.S.; Fréty, R.; Pacheco, J.G.A. Influence of Ni/Al Ratio on the Fast Pyrolysis of Myristic Acid When Adsorbed on Unsupported Mixed Oxides Derived from Layered Double Hydroxides. Catal. Today 2021, 381, 181–191. [Google Scholar] [CrossRef]
- Santos, M.R.; Sales, R.F.; Silva, A.O.S.; Teixeira, C.M.; Pacheco, J.G.A.; Fréty, R. Flash Pyrolysis of Myristic Acid Adsorbed on Supported Nickel Catalysts for Biofuel Production. J. Therm. Anal. Calorim. 2015, 119, 1875–1885. [Google Scholar] [CrossRef]
- Hongloi, N.; Prapainainar, P.; Prapainainar, C. Review of Green Diesel Production from Fatty Acid Deoxygenation over Ni-Based Catalysts. Mol. Catal. 2022, 523, 111696. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, H.; Wang, C.; Chen, K.; Lu, X.; Ouyang, P.; Fu, J. Efficient and Stable Cu–Ni/ZrO2 Catalysts for in Situ Hydrogenation and Deoxygenation of Oleic Acid into Heptadecane Using Methanol as a Hydrogen Donor. Fuel 2018, 230, 211–217. [Google Scholar] [CrossRef]
- Caldeira, V.P.S.; Santos, A.G.D.; Oliveira, D.S.; Lima, R.B.; Souza, L.D.; Pergher, S.B.C. Polyethylene Catalytic Cracking by Thermogravimetric Analysis: Effects of Zeolitic Properties and Homogenization Process. J. Therm. Anal. Calorim. 2017, 130, 1939–1951. [Google Scholar] [CrossRef]
- Araújo, A.M.M.; Queiroz, G.S.M.; Maia, D.O.; Gondim, A.D.; Souza, L.D.; Fernandes, V.J.; Araujo, A.S. Fast Pyrolysis of Sunflower Oil in the Presence of Microporous and Mesoporous Materials for Production of Bio-Oil. Catalysts 2018, 8, 261. [Google Scholar] [CrossRef]
- Qazi, U.Y.; Javaid, R.; Ikhlaq, A.; Khoja, A.H.; Saleem, F. A Comprehensive Review on Zeolite Chemistry for Catalytic Conversion of Biomass/Waste into Green Fuels. Molecules 2022, 27, 8578. [Google Scholar] [CrossRef]
- Zheng, Z.; Lei, T.; Wang, J.; Wei, Y.; Liu, X.; Yu, F.; Ji, J. Catalytic cracking of soybean oil for biofuel over γ-Al2O3/CaO composite catalyst. J. Braz. Chem. Soc. 2019, 30, 359–370. [Google Scholar] [CrossRef]
- Yan, K.; Liu, Y.; Lu, Y.; Chai, J.; Sun, L. Catalytic Application of Layered Double Hydroxide-Derived Catalysts for the Conversion of Biomass-Derived Molecules. Catal. Sci. Technol. 2017, 7, 1622–1645. [Google Scholar] [CrossRef]
- Coriolano, A.C.F.; Alves, A.A.; Araujo, R.A.; Delgado, R.C.; Carvalho, F.R.; Fernandes, V.J.; Araujo, A.S. Thermogravimetry study of the ester interchange of sunflower oil using Mg/Al layered double hydroxides (LDH) impregnated with potassium. J. Therm. Anal. Calorim. 2017, 127, 1863–1867. [Google Scholar] [CrossRef]
- Aguilera, L.J.; Palacio, L.A.; Faro, A.C. Synthesis of NiAl Layered Double Hydroxides Intercalated with Aliphatic Dibasic Anions and Their Exchange with Heptamolybdate. Appl. Clay Sci. 2019, 176, 29–37. [Google Scholar] [CrossRef]
- Tajuddin, N.A.; Manayil, J.C.; Lee, A.F.; Wilson, K. Alkali-free hydrothermally reconstructed NiAl layered double hydroxides for catalytic transesterification. Catalysts 2022, 12, 286. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirb, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Lopes, D.; Zotin, F.; Palacio, L.A. Copper-Nickel Catalysts Hydrotalcite from Hydrotalcite Precursors: The Performance in NO Reduction by CO. Appl. Catal. B 2018, 237, 327–338. [Google Scholar] [CrossRef]
- Evans, D.G.; Slade, R.C.T. Structural Aspects of Layered Double Hydroxides. Struct. Bond. 2005, 119, 1–87. [Google Scholar] [CrossRef]
- Miyata, S. The Syntheses of Hydrotalcite-Like Compounds and their Structures and Physico-Chemical Properties I: The Systems Mg2+-Al3+-NO3−, Mg2+-Al3+-Cl−, Mg2+-Al3+-ClO4−, Ni2+-Al3+-Cl− and Zn2+-Al3+-Cl−. Clays Clay Miner. 1975, 23, 369–375. [Google Scholar] [CrossRef]
- Kooli, F.; Chisem, I.C.; Vucelic, M.; Jones, W. Synthesis and Properties of Terephthalate and Benzoate Intercalates of Mg-Al Layered Double Hydroxides Possessing Varying Layer Charge. Chem. Mater. 1996, 8, 1969–1977. [Google Scholar] [CrossRef]
- Rodrigues, R.M.L.; Reis, C.B.; Eon, J.G.; Palacio, L.A. Oxidative Dehydrogenation of Propane Using Ni–Mg–Al Mixed Oxide Catalysts from Hydrotalcite-Type Precursors. Braz. J. Chem. Eng. 2022. [Google Scholar] [CrossRef]
- Newman, S.P.; Jones, W. Synthesis, Characterization and Applications of Layered Double Hydroxides Containing Organic Guests. New J. Chem. 1998, 22, 105–115. [Google Scholar] [CrossRef]
- Kim, S.C. The Catalytic Oxidation of Aromatic Hydrocarbons over Supported Metal Oxide. J. Hazard. Mater 2002, 91, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Ashok, J.; Subrahmanyam, M.; Venugopal, A. Hydrotalcite Structure Derived Ni-Cu-Al Catalysts for the Production of H2 by CH4 Decomposition. Int. J. Hydrog. Energy 2008, 33, 2704–2713. [Google Scholar] [CrossRef]
- Luo, L.; Yuan, F.; Zaera, F.; Zhu, Y. Catalytic Hydrogenation of Furfural to Furfuryl Alcohol on Hydrotalcite-Derived CuxNi3−xAlOy Mixed-Metal Oxides. J. Catal. 2021, 404, 420–429. [Google Scholar] [CrossRef]
- Lin, X.; Zhu, H.; Huang, M.; Wan, C.; Li, D.; Jiang, L. Controlled Preparation of Ni–Cu Alloy Catalyst via Hydrotalcite-like Precursor and Its Enhanced Catalytic Performance for Methane Decomposition. Fuel Process. Technol. 2022, 233, 107271. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Khzouz, M.; Gkanas, E.I. Experimental and Numerical Study of Low Temperature Methane Steam Reforming for Hydrogen Production. Catalysts 2018, 8, 5. [Google Scholar] [CrossRef]
- Yaakob, Z.; Bshish, A.; Ebshish, A.; Tasirin, S.M.; Alhasan, F.H. Hydrogen Production by Steam Reforming of Ethanol over Nickel Catalysts Supported on Sol Gel Made Alumina: Influence of Calcination Temperature on Supports. Materials 2013, 6, 2229–2239. [Google Scholar] [CrossRef]
- Dragoi, B.; Ungureanu, A.; Chirieac, A.; Ciotonea, C.; Rudolf, C.; Royer, S.; Dumitriu, E. Structural and Catalytic Properties of Mono- and Bimetallic Nickel-Copper Nanoparticles Derived from MgNi(Cu)Al-LDHs under Reductive Conditions. Appl. Catal. A Gen. 2015, 504, 92–102. [Google Scholar] [CrossRef]
- Santos, M.R.; Arias, S.; Padilha, J.F.; Carneiro, M.C.N.; Sales, E.A.; Pacheco, J.G.A.; Fréty, R. Catalytic Cracking of Palmitic and Oleic Acids Pre-Adsorbed on γ-Alumina. Catal. Today 2020, 344, 234–239. [Google Scholar] [CrossRef]
- Maher, K.D.; Bressler, D.C. Pyrolysis of Triglyceride Materials for the Production of Renewable Fuels and Chemicals. Bioresour. Technol. 2007, 98, 2351–2368. [Google Scholar] [CrossRef] [PubMed]
- Krobkrong, N.; Itthibenchapong, V.; Khongpracha, P.; Faungnawakij, K. Deoxygenation of Oleic Acid under an Inert Atmosphere Using Molybdenum Oxide-Based Catalysts. Energy Convers. Manag. 2018, 167, 1–8. [Google Scholar] [CrossRef]
- Padilha, J.F.; Frety, R.; Santos, A.P.; Pontes, L.A.M.; Santos, M.R.; Arias, S.; Pacheco, J.G.A. Deoxygenation of Oleic Acid Methyl Ester in FCC Process Conditions Over Protonated and Sodium Exchanged Y and ZSM-5 Zeolites. Waste Biomass Valorization 2022, 13, 185–194. [Google Scholar] [CrossRef]
- Asomaning, J.; Mussone, P.; Bressler, D.C. Pyrolysis of polyunsaturated fatty acids. Fuel Process. Technol. 2014, 120, 89–95. [Google Scholar] [CrossRef]
- Lischka, H.; Ventura, E.; Dallos, M. The Diels-Alder Reaction of Ethene and 1,3-Butadiene: An Extended Multireference Ab Initio Investigation. ChemPhysChem 2004, 5, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Kubátová, A.; Št’ávová, J.; Seames, W.S.; Luo, Y.; Sadrameli, S.M.; Linnen, M.J.; Baglayeva, G.V.; Smoliakova, I.P.; Kozliak, E.I. Triacylglyceride Thermal Cracking: Pathways to Cyclic Hydrocarbons. Energy Fuels 2012, 26, 672–685. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Liu, C.; Lu, Y.; Lin, X.; Hou, D.; Luo, C.; Wang, D.; Zheng, Z.; Zheng, Y. Highly Stable Mo-Based Bimetallic Catalysts for Selective Deoxygenation of Oleic Acid to Fuel-like Hydrocarbons. J. Environ. Chem. Eng. 2023, 11, 109104. [Google Scholar] [CrossRef]
- Wang, H.; Lin, H.; Zheng, Y.; Ng, S.; Brown, H.; Xia, Y. Kaolin-Based Catalyst as a Triglyceride FCC Upgrading Catalyst with High Deoxygenation, Mild Cracking, and Low Dehydrogenation Performances. Catal. Today 2019, 319, 164–171. [Google Scholar] [CrossRef]
- Fu, L.; Li, Y.; Cui, H.; Ba, W.; Liu, Y. Highly Stable and Selective Catalytic Deoxygenation of Renewable Bio-Lipids over Ni/CeO2-Al2O3 for N-Alkanes. Appl. Catal. A Gen. 2021, 623, 118258. [Google Scholar] [CrossRef]
- Billaud, F.; Minh, A.K.T.; Lozano, P.; Pioch, D. Catalytic Cracking of Octanoic Acid. J. Anal. Appl. Pyrolysis 2001, 58–59, 605–616. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ni (%) | Cu (%) | Al (%) | Theoretical Molar Ratios | Experimental Molar Ratios | ||
|---|---|---|---|---|---|---|---|
| x | Cu/Ni | x | Cu/Ni | ||||
| NiAl | 60.3 | --- | 39.7 | 0.70 | --- | 0.59 | --- |
| 0.1CuNiAl | 55.3 | 6.5 | 38.2 | 0.70 | 0.10 | 0.58 | 0.11 |
| 0.2CuNiAl | 50.5 | 11.5 | 38.0 | 0.70 | 0.20 | 0.57 | 0.21 |
| 0.4CuNiAl | 44.1 | 20.5 | 35.3 | 0.70 | 0.40 | 0.55 | 0.43 |
| Sample | Surface Area (m2 g−1) a | Average Pore Diameter (Å) b | Pore Volume (cm3 g−1) b | NiAl2O4 Average Crystallite Size (nm) c |
|---|---|---|---|---|
| NiAl | 230 | 100 | 0.65 | 2.9 |
| 0.1CuNiAl | 272 | 85 | 0.67 | 2.7 |
| 0.2CuNiAl | 253 | 77 | 0.54 | 2.4 |
| 0.4CuNiAl | 262 | 55 | 0.40 | 2.9 |
| Catalyst | Linear Alkanes | Linear Alkenes | Cycloalkene | Ramified Alkenes |
|---|---|---|---|---|
| Pure Oleic Acid | 1.12 | 20.63 | 6.28 | --- |
| NiAl | 4.21 | 40.67 | 2.29 | 0.82 |
| 0.1CuNiAl | 5.45 | 39.92 | 3.24 | 1.33 |
| 0.2CuNiAl | 5.52 | 42.15 | 3.29 | 1.22 |
| 0.4CuNiAl | 7.01 | 42.96 | 3.49 | 0.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabino, J.; Liborio, D.O.; Arias, S.; Gonzalez, J.F.; Barbosa, C.M.B.M.; Carvalho, F.R.; Frety, R.; Barros, I.C.L.; Pacheco, J.G.A. Hydrogen-Free Deoxygenation of Oleic Acid and Industrial Vegetable Oil Waste on CuNiAl Catalysts for Biofuel Production. Energies 2023, 16, 6131. https://doi.org/10.3390/en16176131
Sabino J, Liborio DO, Arias S, Gonzalez JF, Barbosa CMBM, Carvalho FR, Frety R, Barros ICL, Pacheco JGA. Hydrogen-Free Deoxygenation of Oleic Acid and Industrial Vegetable Oil Waste on CuNiAl Catalysts for Biofuel Production. Energies. 2023; 16(17):6131. https://doi.org/10.3390/en16176131
Chicago/Turabian StyleSabino, Jose, Denisson O. Liborio, Santiago Arias, Juan F. Gonzalez, Celmy M. B. M. Barbosa, Florival R. Carvalho, Roger Frety, Ivoneide C. L. Barros, and Jose Geraldo A. Pacheco. 2023. "Hydrogen-Free Deoxygenation of Oleic Acid and Industrial Vegetable Oil Waste on CuNiAl Catalysts for Biofuel Production" Energies 16, no. 17: 6131. https://doi.org/10.3390/en16176131
APA StyleSabino, J., Liborio, D. O., Arias, S., Gonzalez, J. F., Barbosa, C. M. B. M., Carvalho, F. R., Frety, R., Barros, I. C. L., & Pacheco, J. G. A. (2023). Hydrogen-Free Deoxygenation of Oleic Acid and Industrial Vegetable Oil Waste on CuNiAl Catalysts for Biofuel Production. Energies, 16(17), 6131. https://doi.org/10.3390/en16176131