Lithium-ion (Li-ion) batteries are the predominant energy storage and conversion device in various applications, such as portable consumer electronics, electric vehicles (EVs), grid storage, space applications, and military applications [
1,
2,
3]. Li-ion batteries offer several advantages over other rechargeable batteries, such as high energy density and power density, low self-discharge rate, and long life [
4]. However, decomposition reactions occur within Li-ion batteries under certain operating conditions, which negatively impact the performance of the battery and impose safety hazards by generating toxic and flammable compounds. For instance, under abuse conditions, decomposition reactions generate a significant amount of heat and gaseous products, leading the cell into thermal runaway. Thermal runaway is frequently accompanied by the release of flammable and toxic gases during the venting process and afterward. The rates and pathways of these decomposition reactions, the composition and amount of the gaseous products, and heat generation rates are affected by the solvent(s) used within the electrolyte solution, which can vary significantly [
5]. The most common electrolytes used in Li-ion batteries comprise a conducting salt such as lithium hexafluorophosphate (LiPF
6) dissolved in a mixture of organic carbonate solvents.
Accurate analytical methods for determining the composition of solvent systems used in Li-ion batteries are needed to better understand decomposition processes occurring within Li-ion batteries and their effects on battery performance and safety. Recovering organic carbonate compounds during recycling may also benefit from accurate analytical methods to verify the solvent composition [
6]. Several studies have implemented different analytical methods to identify and quantify electrolyte components. For example, in a qualitative study, Horsthemke et al. [
7] used gas chromatography combined with mass spectrometry (GC/MS) to identify electrolyte components and aging products for various commercially available Li-ion cells. A headspace solid-phase microextraction (SPME) technique was used to prevent the injection of salts into the GC column [
7]. On the other hand, Ellis et al. [
8] have used Fourier-transform infrared (FTIR) spectroscopy to determine the concentrations of LiPF
6 and common solvents in Li-ion battery electrolytes. An FTIR spectral database of known solutions with known concentrations was first created. Machine learning was then used to quantify unknown electrolyte solutions by interpolating data in the FTIR spectral database [
8]. Further, Gachot et al. [
9] have used the GC/MS technique to identify the compounds within the Li-ion battery electrolytes after cycling and heating. A postcapillary column was used to preserve the ion source from HF and the bleeding of the first column. The same research group has coupled gas chromatography with mass spectrometry and Fourier transform infrared spectroscopy (GC/MS-FTIR) to identify electrolyte compositions. This method was used to characterize volatile compounds released during thermal runaway and gaseous and soluble volatile products in a swollen battery [
10]. Schultz et al. [
11] have employed high-performance liquid chromatography (HPLC) coupled to tandem mass spectrometry (LC/MS/MS) for the identification and quantification of components of Li-ion battery electrolytes. The method was then used to quantify chemical species produced under different aging processes [
11]. Petibon et al. [
12] have developed a semi-quantitative approach based on the GC/MS technique for the determination of the consumption and transesterification of additives and solvents after formation cycles and storage at high potential. Liquid–liquid extraction was used to remove LiPF
6 prior to analysis [
12]. However, liquid–liquid extraction is time-consuming and may introduce additional uncertainty in the measurement. Terborg et al. [
13] have employed GC/MS and GC/FID to identify organic solvents in Li-ion battery electrolytes. Thompson et al. [
14] have used GC/MS to analyze the changes in the composition of the carbonates within the battery electrolytes after cycling at different cutoff potentials. The salt was extracted from the samples using liquid–liquid extraction and centrifuge [
14]. Weber et al. [
15] have used a GC–MS-based analytical method to identify phosphorous/organic degradation products in aged cells that are known to be toxic. Quantification of dimethyl phosphorofluoridate (DMPF) and diethyl phosphorofluoridate (DEPF) has been done using calibration curves [
15]. Finally, several previous studies have used gas chromatography to investigate venting behavior and venting gas composition of lithium-ion cells during thermal runaway [
10,
16,
17,
18,
19].
Most of the previously reported techniques required removing LiPF
6 salt from the samples prior to analysis because it is well known that the conductive salt reacts with the silicon coating of the GC column and results in excessive column bleeding [
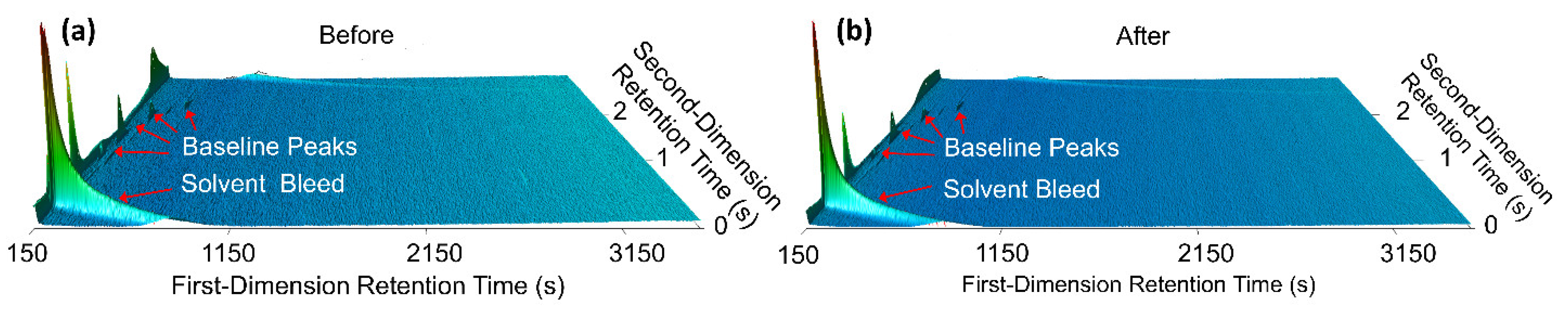
7]. However, salt removal is time-consuming, introduces additional costs, and reduces the precision of the method.
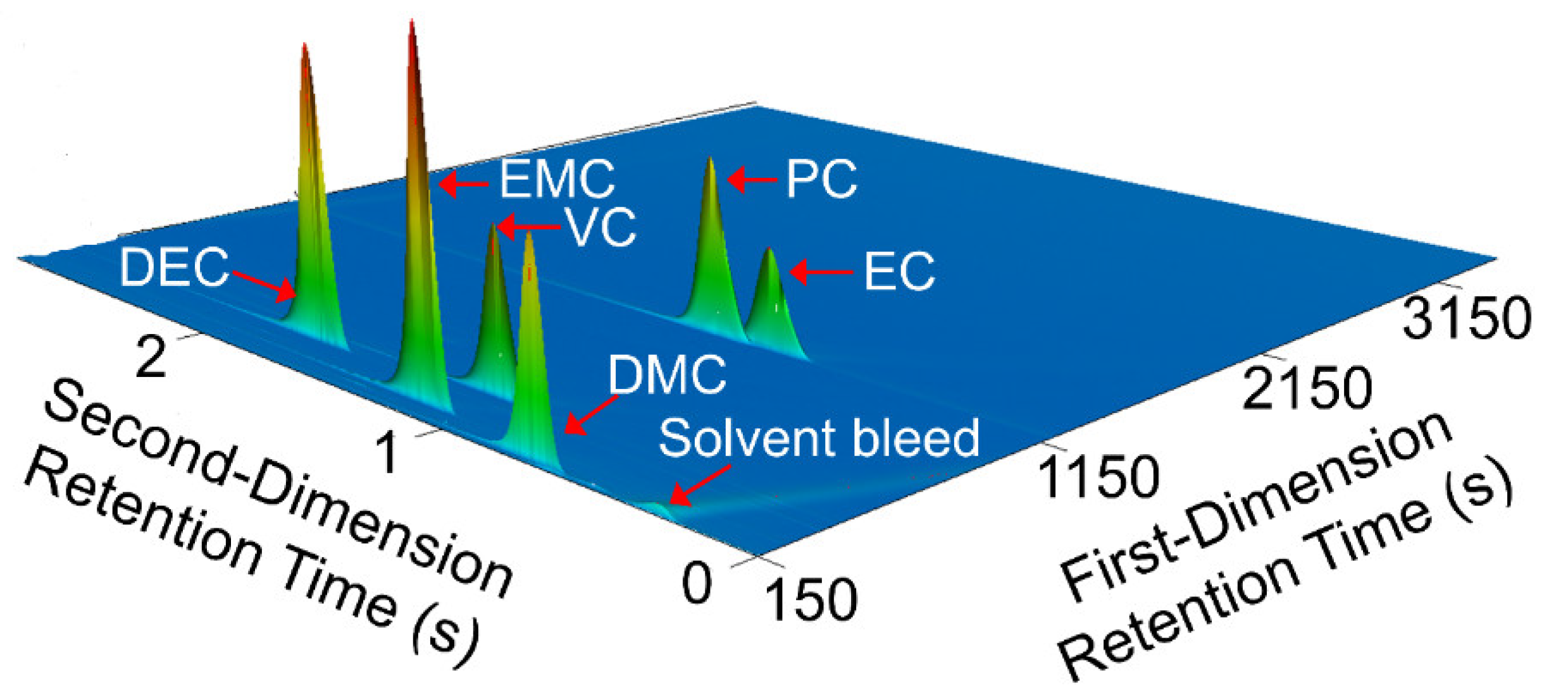
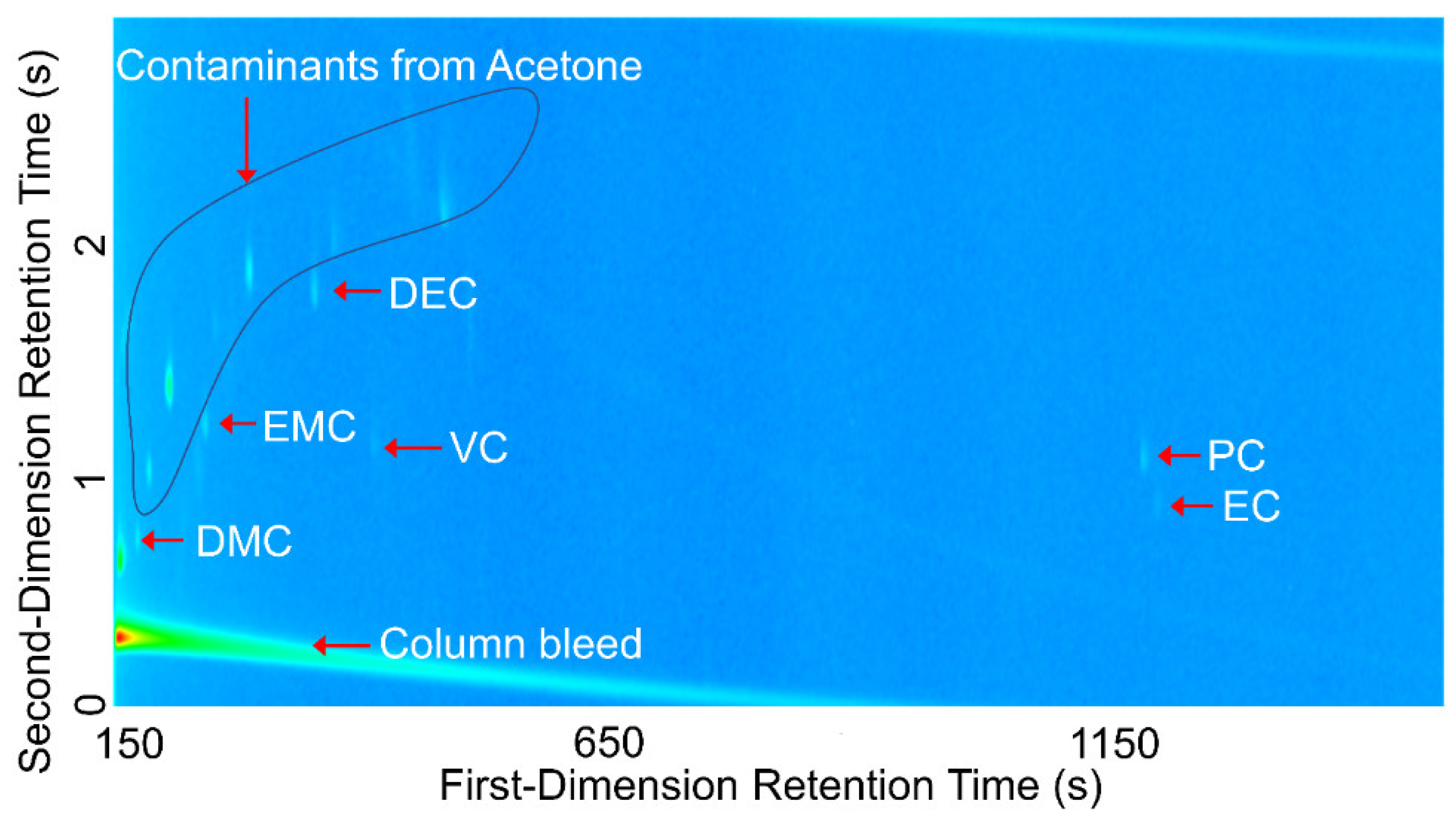
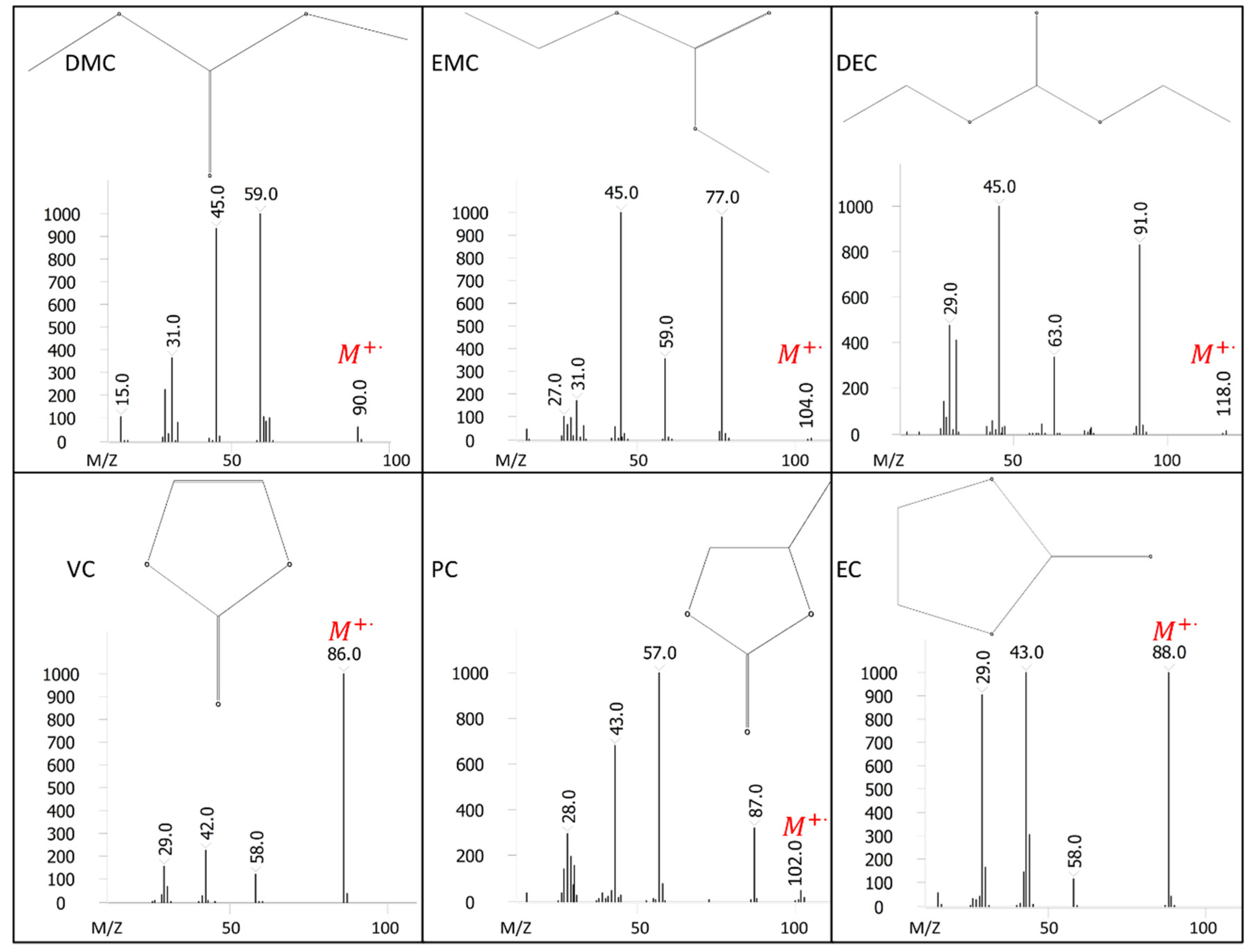
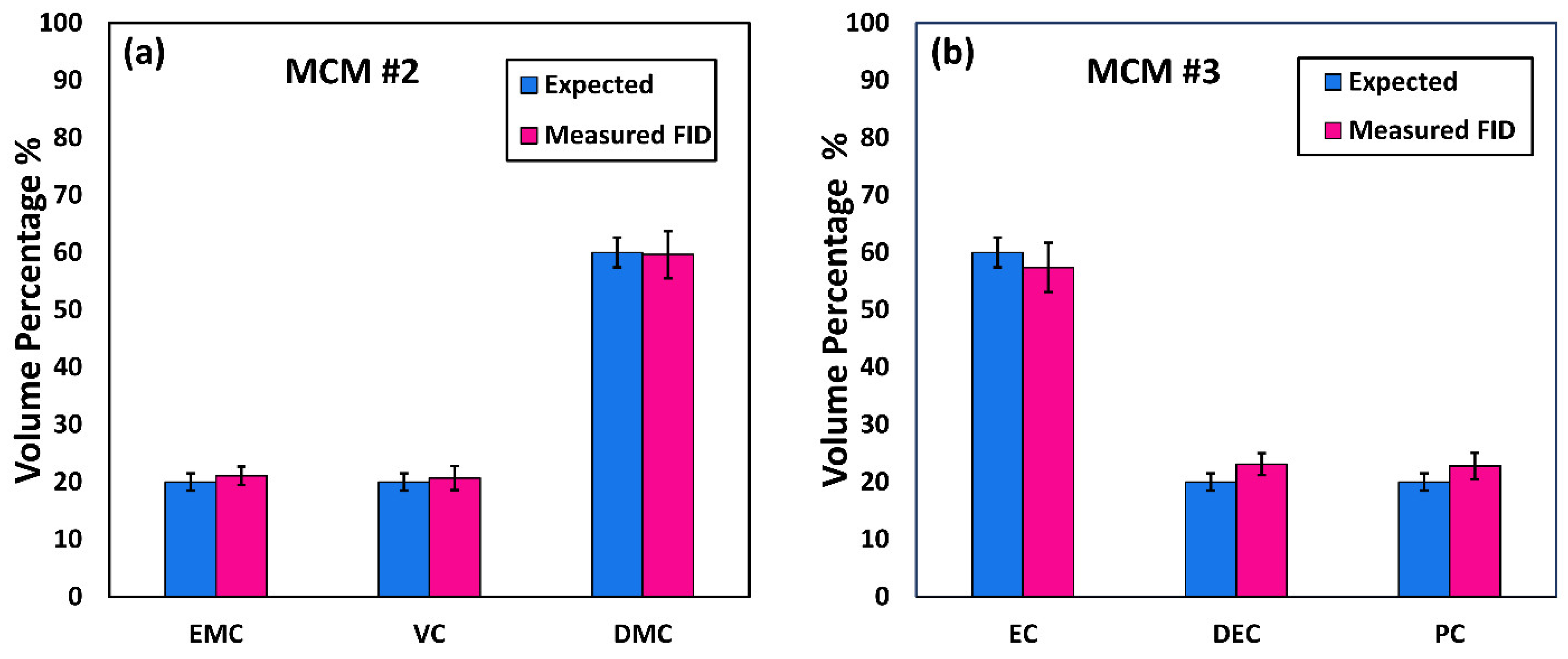
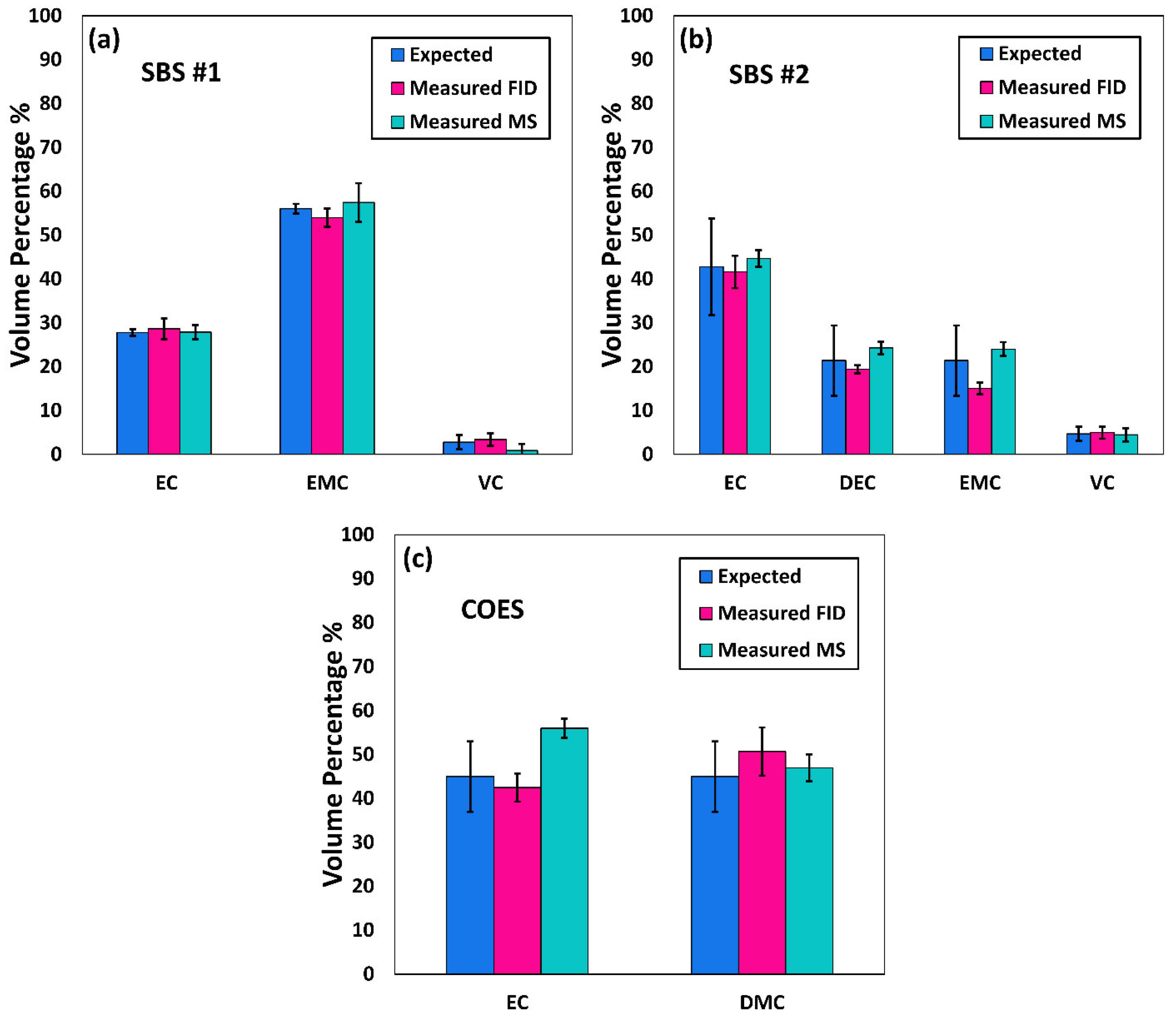
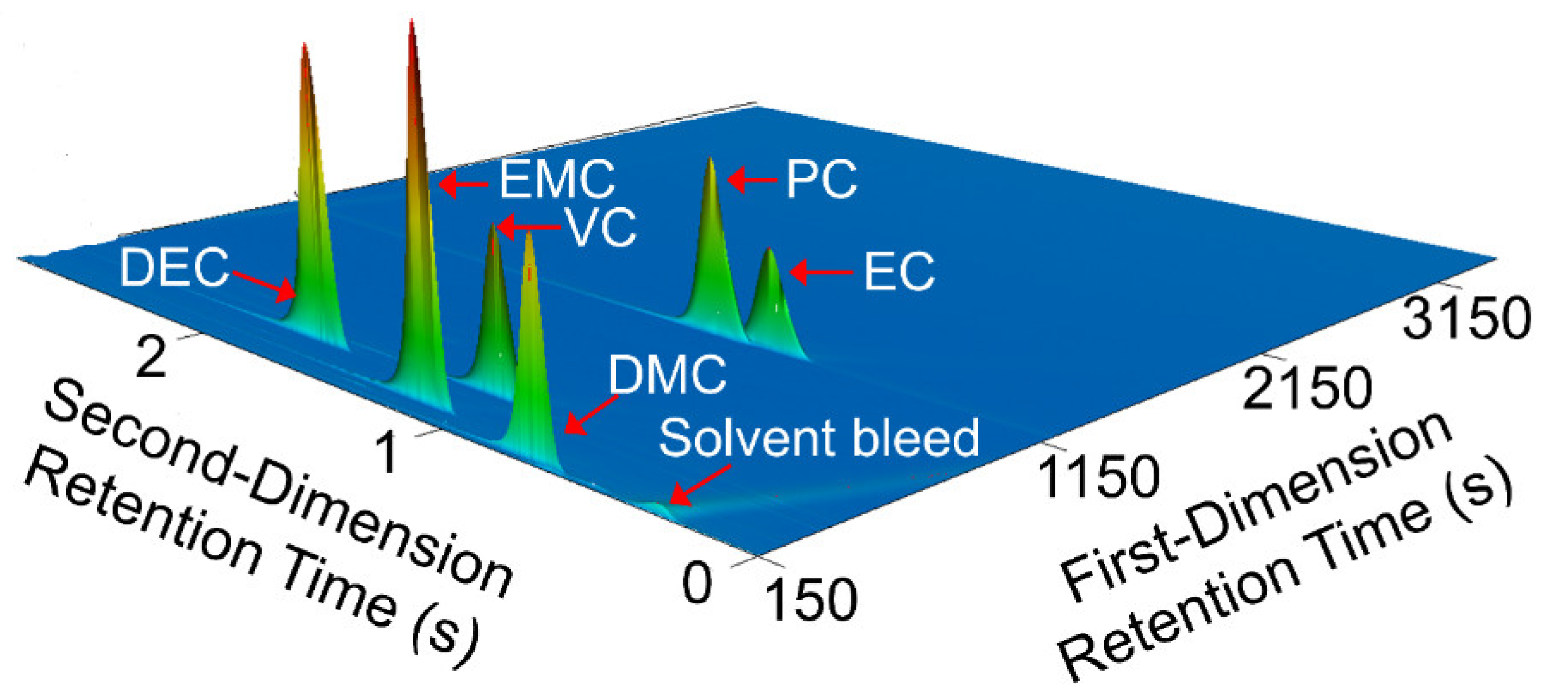
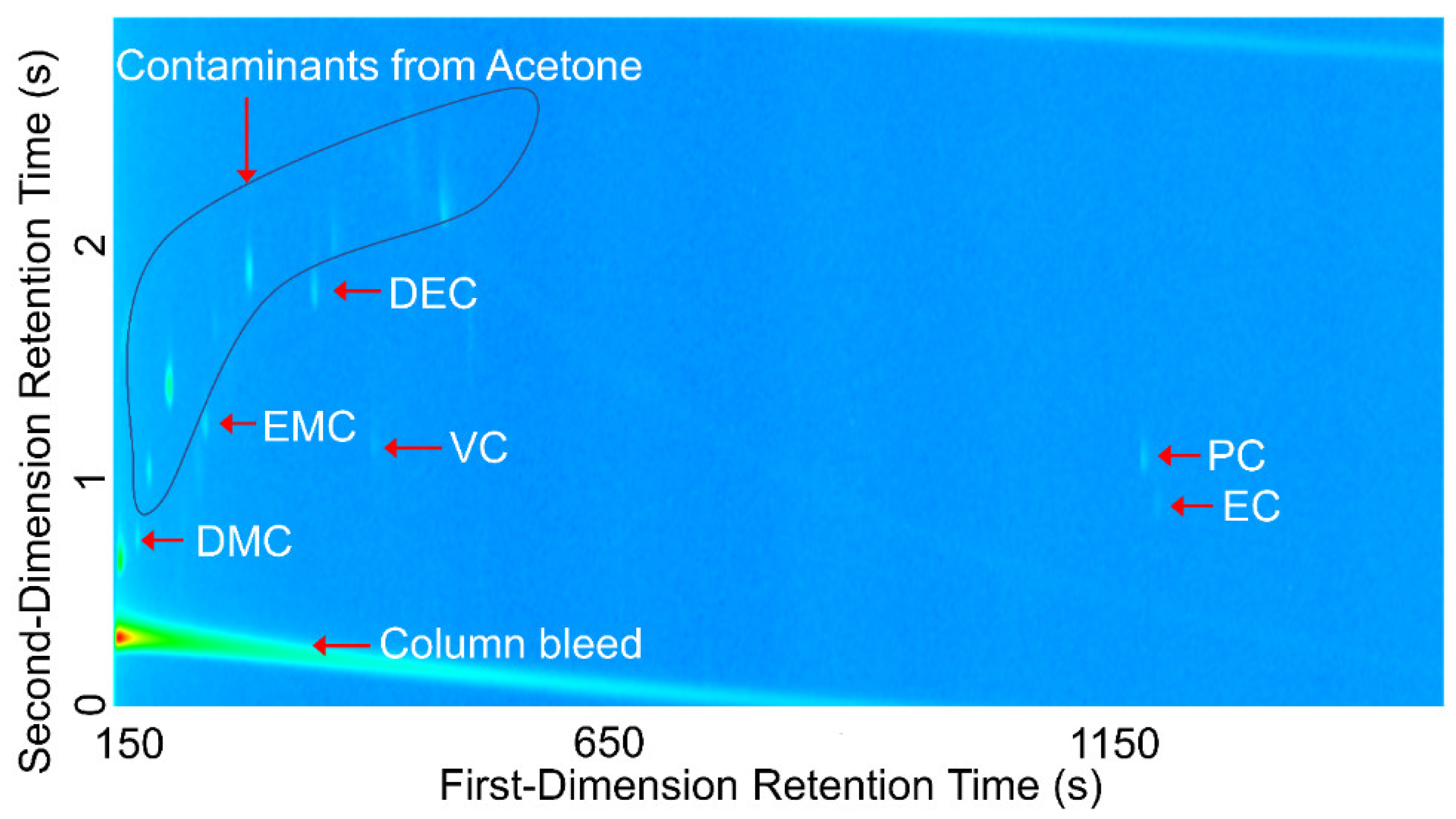
Comprehensive two-dimensional gas chromatography (GC×GC) coupled with FID (GC×GC/FID), or mass spectrometry detection (GC×GC/EI TOF MS) is a powerful analytical technique due to its high resolution and high peak capacity. MS can often be used for the identification of unknown compounds based on a comparison of their electron ionization (EI) mass spectra to mass spectral libraries, while FID is often used for compound quantification because of the similar response of FID to different types of compounds. In this paper, two methods are described for the determination of the compositions of solvent systems used in Li-ion battery electrolytes, one based on GC×GC/FID and the other GC×GC/EI TOF MS. GC×GC has been widely used to analyze complex mixtures related to food, petrochemical, environmental, biomedical, hardware, and software industries [
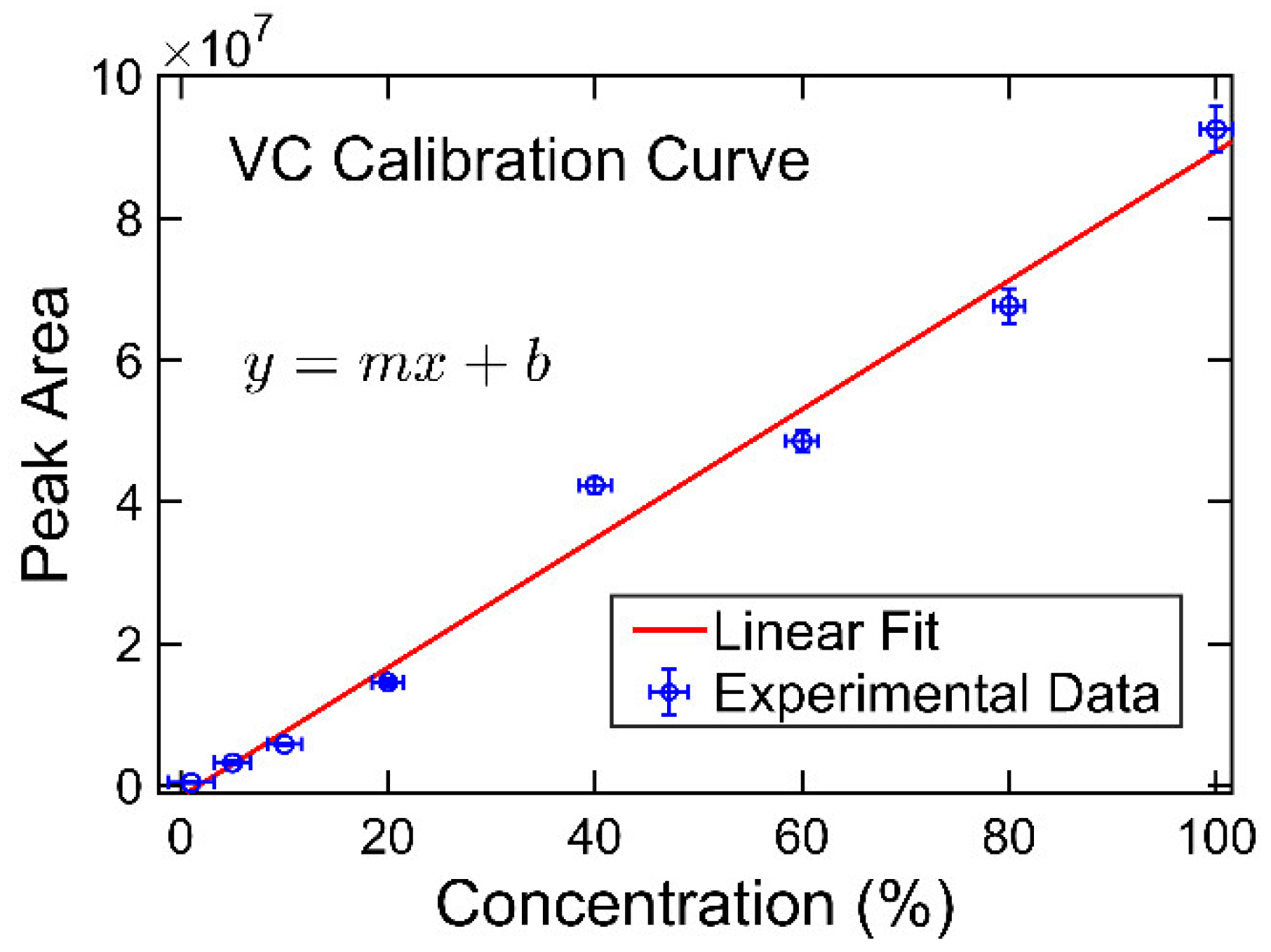
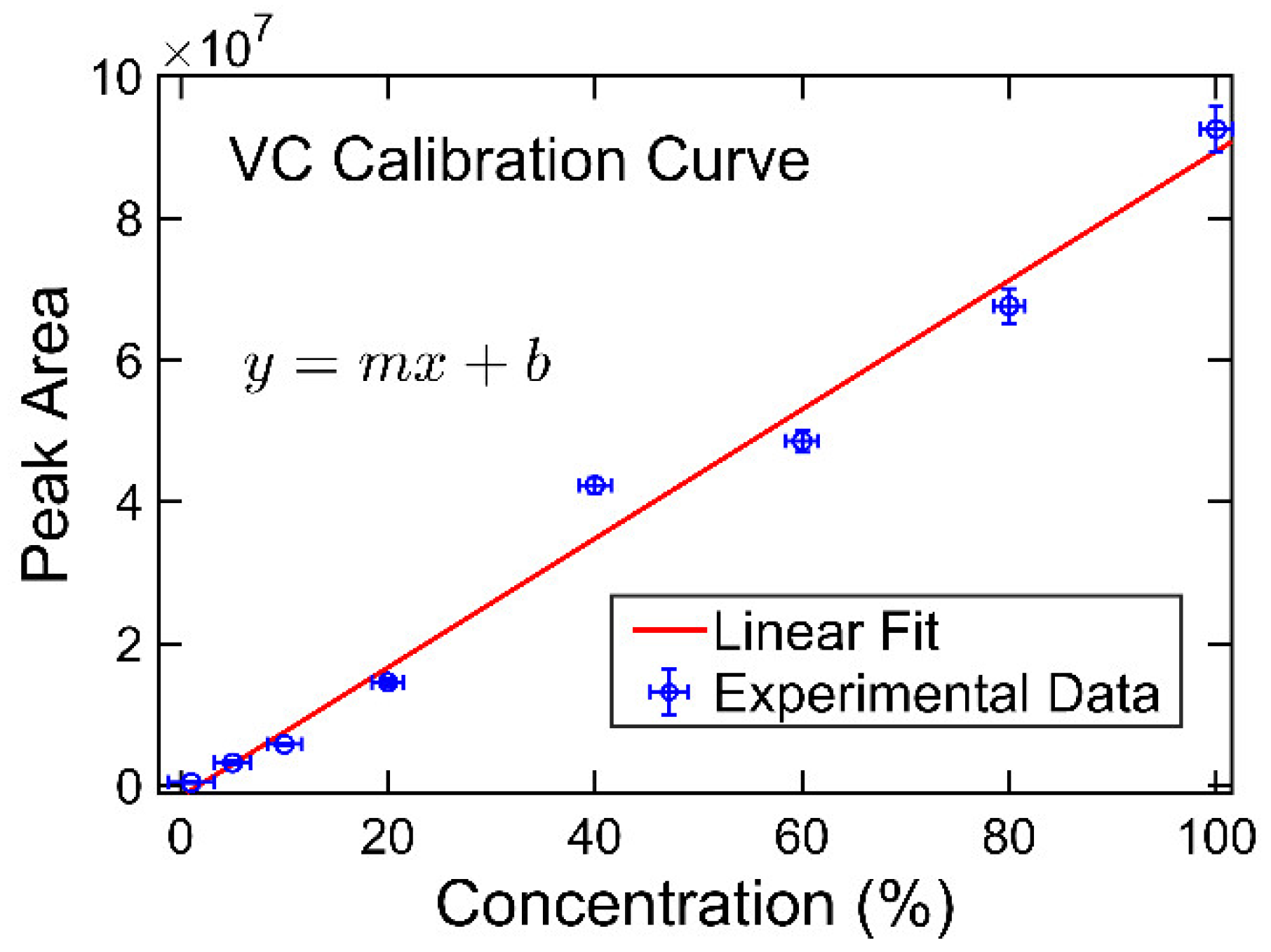
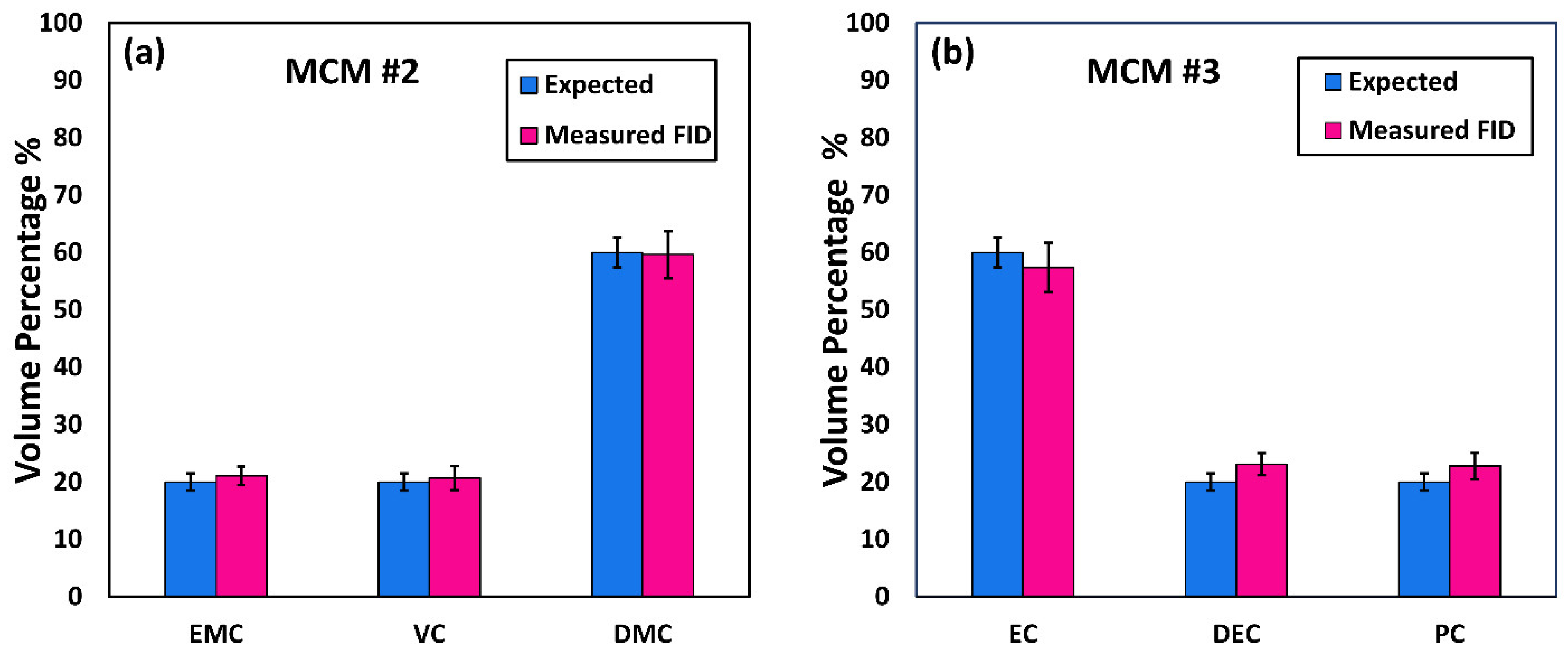
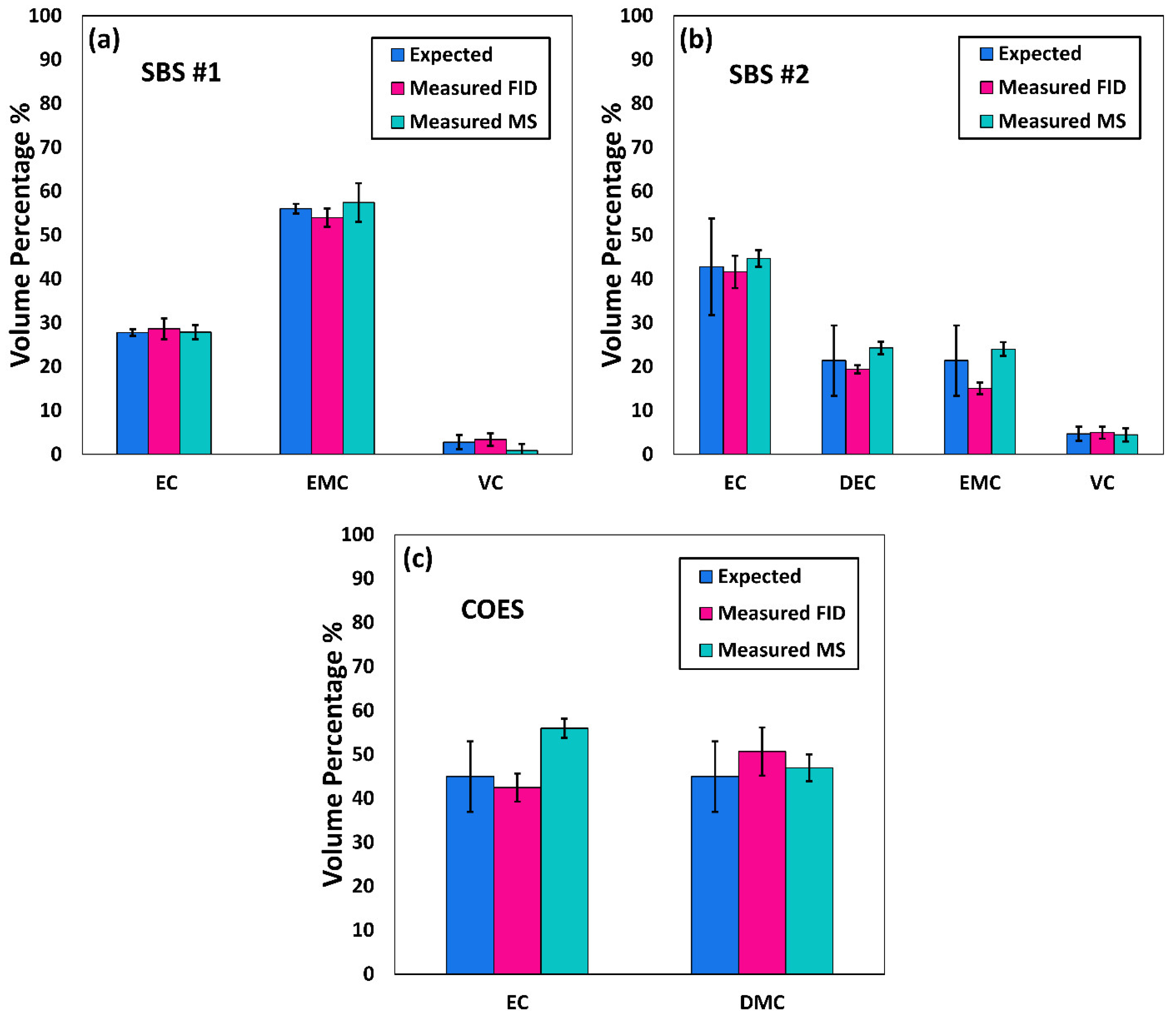
20]. However, to the best of the authors’ knowledge, determination of the compositions of the solvent systems used in Li-ion battery electrolytes has not been performed based on GC×GC/FID or GC×GC/EI TOF MS. The purpose of the present work was to determine the accuracy and precision of these two analytical techniques when analyzing compounds found in electrolyte solutions of Li-ion batteries. To quantify and compare the performance of the two analytical techniques, the limit of detection (LOD), limit of quantification (LOQ), uncertainty, and repeatability were determined.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}