
Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations


Abstract
1. Introduction
2. Computational Method
3. Results and Discussion
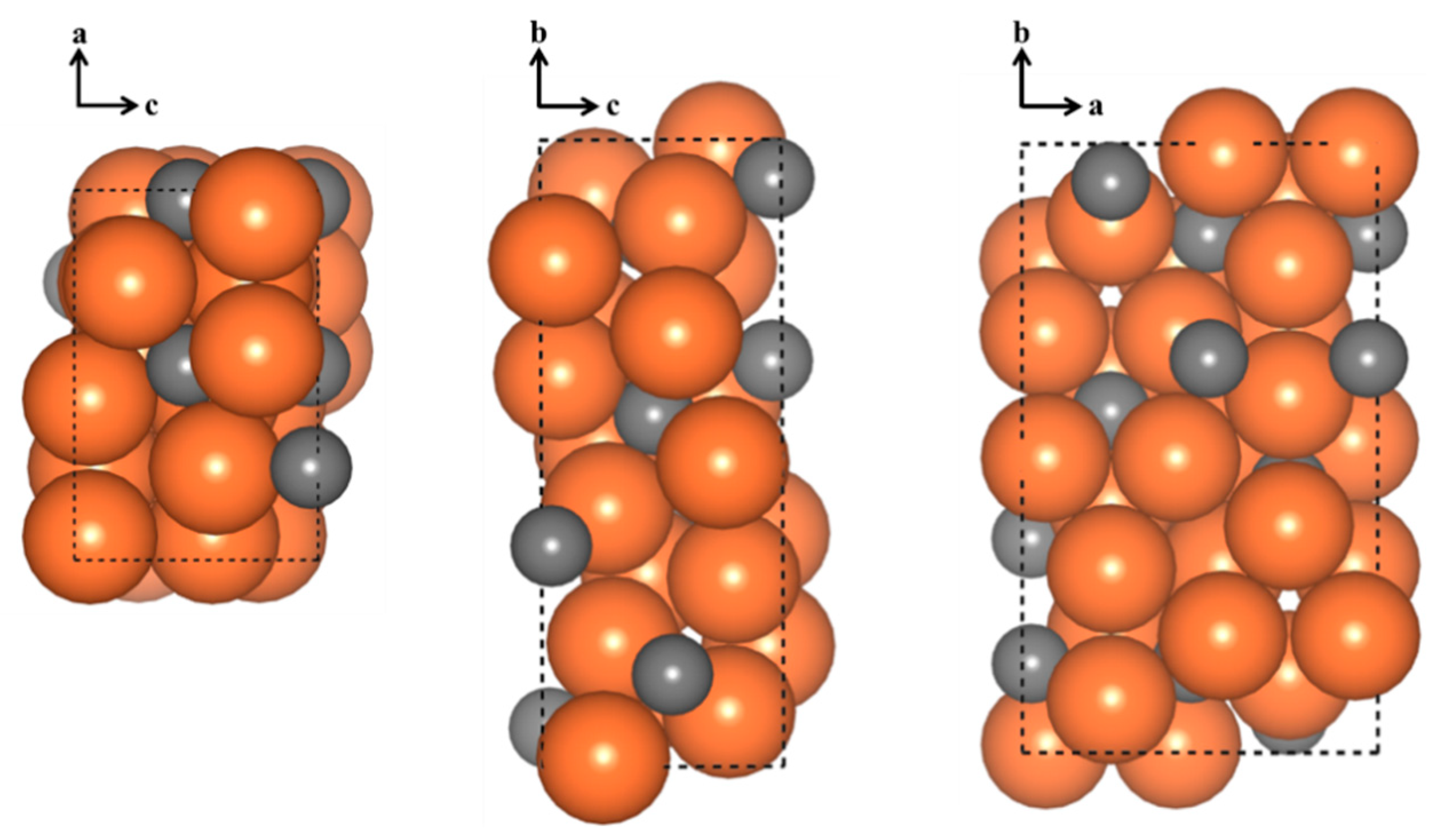
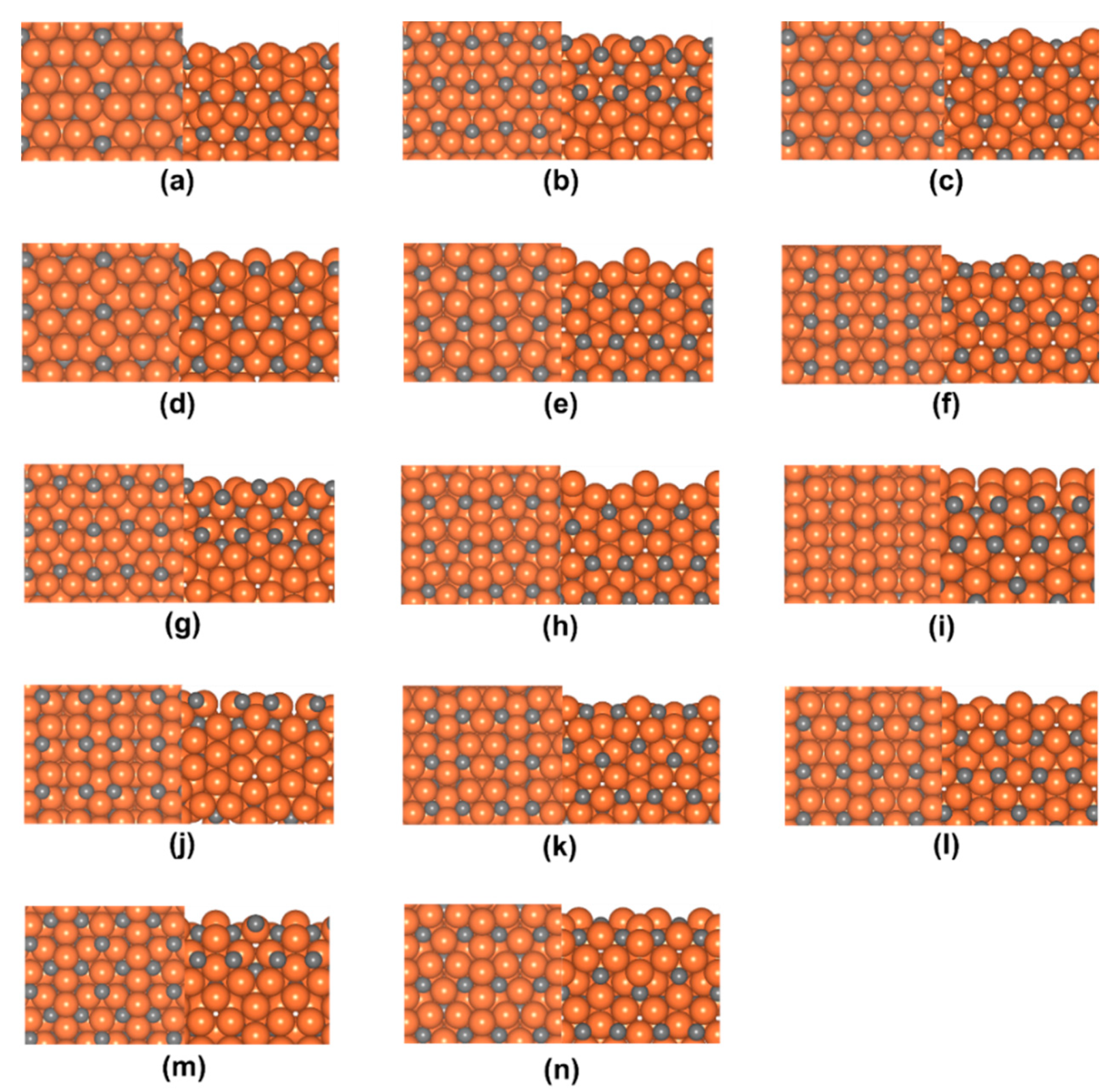
3.1. Surface Energies
3.2. CO* Activation
3.2.1. CO*, C*, and O* Adsorption
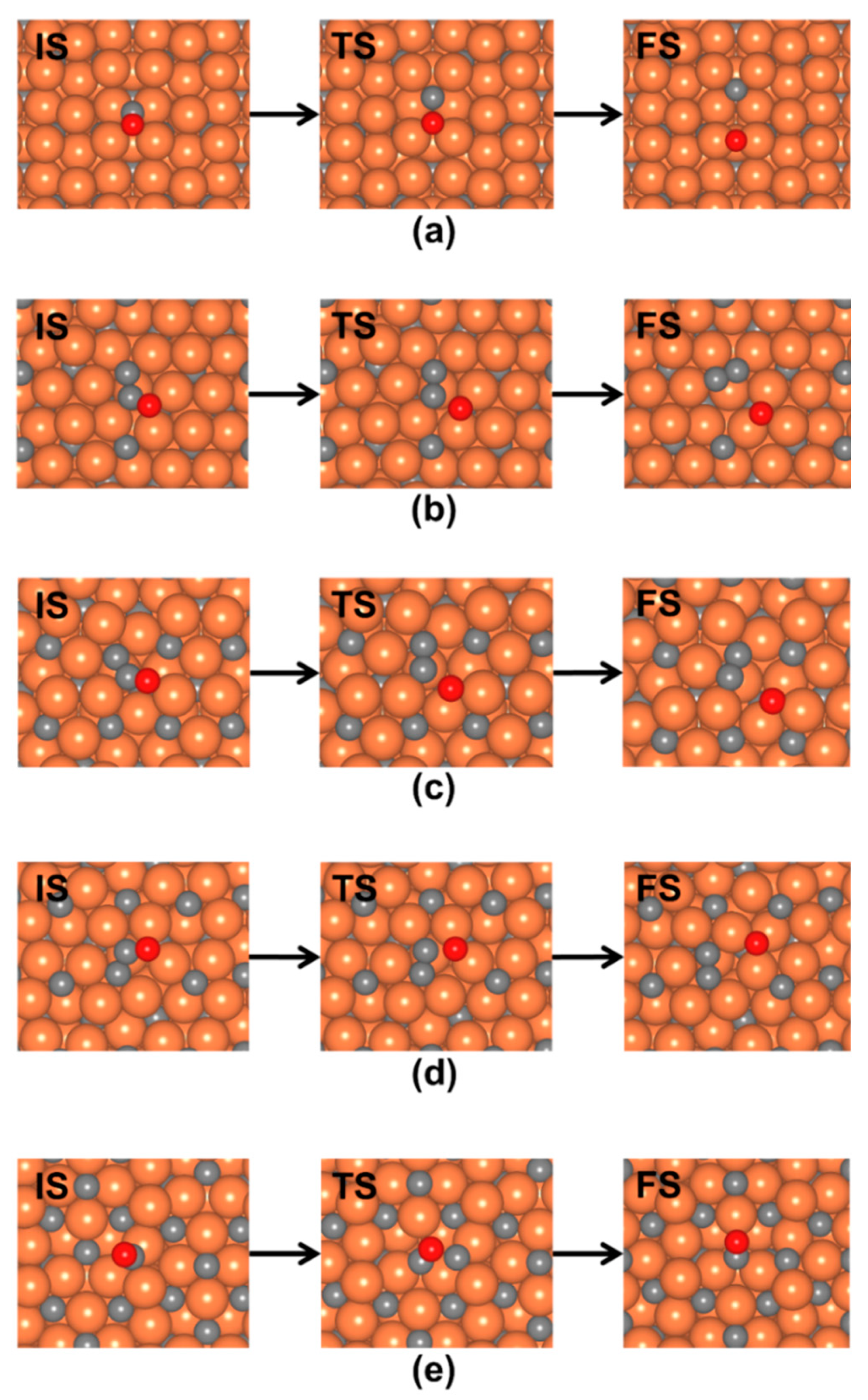
3.2.2. Direct CO* Dissociation
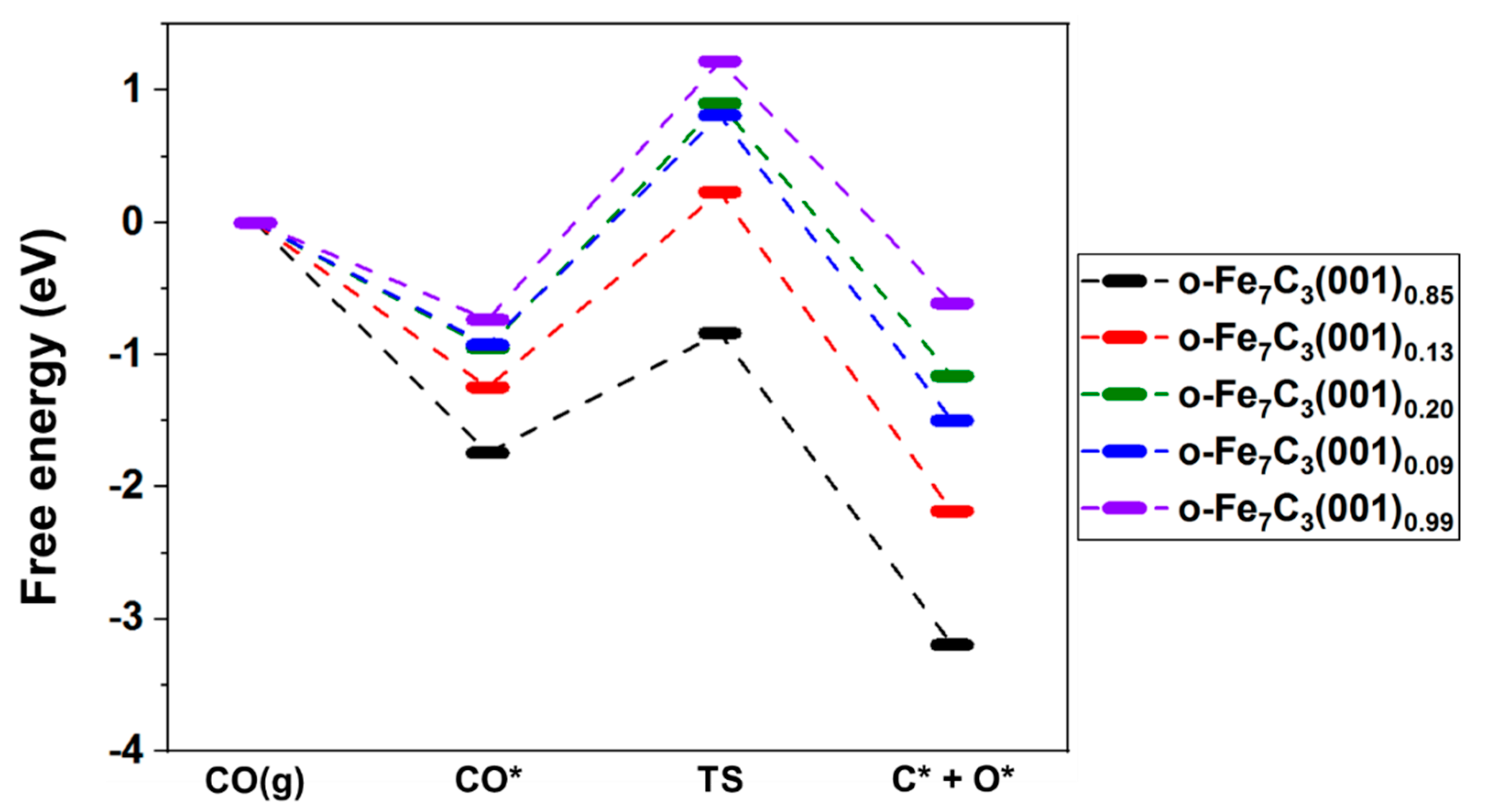
3.2.3. Free Energy Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dry, M.E. The Fischer-Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Steynberg, A.P. Introduction to Fischer-Tropsch Technology. Stud. Surf. Sci. Catal. 2004, 152, 1–63. [Google Scholar]
- Huang, C.-S.; Xu, L.; Davis, B.H. Fischer-Tropsch synthesis: Impact of pretreatment of ultrafine iron oxide upon catalyst structure and selectivity. Fuel Sci. Technol. Int. 1993, 11, 639–664. [Google Scholar] [CrossRef]
- Jin, Y.; Datye, A.K. Phase transformations in iron Fischer-Tropsch catalysts during temperature-programmed reduction. J. Catal. 2000, 196, 8–17. [Google Scholar] [CrossRef]
- Zhang, Q.; Kang, J.; Wang, Y. Development of novel catalysts for Fischer-Tropsch synthesis: Tuning the product selectivity. ChemCatChem 2010, 2, 1030–1058. [Google Scholar] [CrossRef]
- Pham, T.H.; Duan, X.; Qian, G.; Zhou, X.; Chen, D. CO activation pathways of Fischer-Tropsch synthesis on χ-Fe5C2 (510): Direct versus hydrogen-assisted CO dissociation. J. Phys. Chem. C 2014, 118, 10170–10176. [Google Scholar] [CrossRef]
- Chen, B.; Wang, D.; Duan, X.; Liu, W.; Li, Y.; Qian, G.; Yuan, W.; Holmen, A.; Zhou, X.; Chen, D. Charge-tuned CO activation over a χ-Fe5C2 Fischer-Tropsch catalyst. ACS Catal. 2018, 8, 2709–2714. [Google Scholar] [CrossRef]
- Datye, A.K.; Jin, Y.; Mansker, L.; Motjope, R.T.; Dlamini, T.H.; Coville, N.J. The nature of the active phase in iron Fischer-Tropsch catalysts. Stud. Surf. Sci. Catal. 2000, 130, 1139–1144. [Google Scholar]
- Rivera de la Cruz, J.G.; Sabbe, M.K.; Reyniers, M.F. First principle study on the adsorption of hydrocarbon chains involved in Fischer-Tropsch synthesis over iron carbides. J. Phys. Chem. C 2017, 121, 25052–25063. [Google Scholar] [CrossRef]
- Chang, Q.; Zhang, C.; Liu, C.; Wei, Y.; Cheruvathur, A.V.; Dugulan, A.I.; Niemantsverdriet, J.W.; Liu, X.; He, Y.; Qing, M.; et al. Relationship between iron carbide phases (ε-Fe2C, Fe7C3, and χ-Fe5C2) and catalytic performances of Fe/SiO2 Fischer-Tropsch catalysts. ACS Catal. 2018, 8, 3304–3316. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Iglesia, E. CO activation pathways and the mechanism of Fischer-Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Petersen, M.A.; van Rensburg, W.J. CO dissociation at vacancy sites on Hägg iron carbide: Direct versus hydrogen-assisted routes investigated with DFT. Top. Catal. 2015, 58, 665–674. [Google Scholar] [CrossRef]
- Ozbek, M.O.; Niemantsverdriet, J.H. Elementary reactions of CO and H2 on C-terminated χ-Fe5C2(001) surfaces. J. Catal. 2014, 317, 158–166. [Google Scholar] [CrossRef]
- Cao, D.B.; Li, Y.W.; Wang, J.; Jiao, H. Adsorption and reaction of surface carbon species on Fe5C2(001). J. Phys. Chem. C 2008, 112, 14883–14890. [Google Scholar] [CrossRef]
- Yang, S.; Chun, H.-J.; Lee, S.; Han, S.J.; Lee, K.Y.; Kim, Y.T. Comparative study of olefin production from CO and CO2 using Na- and K-promoted zinc ferrite. ACS Catal. 2020, 10, 10742–10759. [Google Scholar] [CrossRef]
- Fang, C.M.; van Huis, M.A.; Zandbergen, H.W. Structural, electronic, and magnetic properties of iron carbide Fe7C3 phases from first-principles theory. Phys. Rev. B 2009, 80, 224108. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G. A climbing image nudged elastic band method for finding saddle points and mini-mum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Zhu, Y.A.; Chen, D.; Zhou, X.G.; Yuan, W.K. DFT studies of dry reforming of methane on Ni catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Cao, X.M.; Burch, R.; Hardacre, C.; Hu, P. An understanding of chemoselective hydrogenation on crotonaldehyde over Pt(111) in the free energy landscape: The microkinetics study based on first-principles calculations. Catal. Today 2011, 165, 71–79. [Google Scholar] [CrossRef]
- Chase, M.W. NIST-JANAF Thermochemical Tables, 4th ed.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1998; p. 643.
- Scholl, D.S.; Steckel, J.A. Density Functional Theory: A Practical Introduction, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 131–161. [Google Scholar]
- Steynberg, P.J.; Van den Berg, J.A.; van Rensburg, W.J. Bulk and surface analysis of Hägg Fe carbide (Fe5C2): A density functional theory study. J. Phys. Condens. Matter 2008, 20, 064238. [Google Scholar] [CrossRef]
- Zhang, J.M.; Pang, Q.; Xu, K.W.; Ji, V. First-principles study of the (001) surface of cubic PbTiO3. Surf. Interface Anal. 2008, 40, 1382–1387. [Google Scholar] [CrossRef]
- Tian, X.; Wang, T.; Fan, L.; Wang, Y.; Lu, H.; Mu, Y. A DFT based method for calculating the surface energies of asymmetric MoP facets. Appl. Surf. Sci. 2018, 427, 357–362. [Google Scholar] [CrossRef]
- García-García, F.R.; Guerrero-Ruiz, A.; Rodriguez-Ramos, I. Role of B5-type sites in Ru catalysts used for the NH3 decomposition reaction. Top. Catal. 2009, 52, 758–764. [Google Scholar] [CrossRef]
- Arevalo, R.L.; Aspera, S.M.; Escaño, M.C.S.; Nakanishi, H.; Kasai, H. First principles study of methane decomposition on B5 step-edge type site of Ru surface. J. Phys. Condens. Matter 2017, 29, 184001. [Google Scholar] [CrossRef]
- Ge, Q.; Neurock, M. Structure dependence of NO adsorption and dissociation on platinum surfaces. J. Am. Chem. Soc. 2004, 126, 1551–1559. [Google Scholar] [CrossRef]
- Rempel, J.; Greeley, J.; Hansen, L.B.; Nielsen, O.H.; Nørskov, J.K.; Mavrikakis, M. Step effects on the dissociation of NO on close-packed rhodium surfaces. J. Phys. Chem. C 2009, 113, 20623–20631. [Google Scholar] [CrossRef]
- Broos, R.J.; Zijlstra, B.; Filot, I.A.; Hensen, E.J. Quantum-chemical DFT study of direct and H- and C-assisted CO dissociation on the χ-Fe5C2 Hägg carbide. J. Phys. Chem. C 2018, 122, 9929–9938. [Google Scholar] [CrossRef] [PubMed]
- Ozbek, M.O.; Niemantsverdriet, J.H. Methane, formaldehyde and methanol formation pathways from carbon monoxide and hydrogen on the (001) surface of the iron carbide χ-Fe5C2. J. Catal. 2015, 325, 9–18. [Google Scholar] [CrossRef]
- Chen, W.; Lin, T.; Dai, Y.; An, Y.; Yu, F.; Zhong, L.; Li, S.; Sun, Y. Recent advances in the investigation of nanoeffects of Fischer-Tropsch catalysts. Catal. Today 2018, 311, 8–22. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I. Systematic trends in (0 0 1) surface ab initio calculations of ABO3 perovskites. J. Saudi Chem. Soc. 2018, 22, 459–468. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Gabrusenoks, J.; Popov, A.; Jia, R. Comparative ab initio calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces. Crystals 2020, 10, 745. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Surface Termination | Surface Energy (eV·Å−2) | Surface C Atom Coverage |
|---|---|---|---|
| 1 | (001)0.20 | 0.140 (2.247) a | 0.33 |
| 2 b | (001)0.09 | 0.153 (2.456) | 0.33 |
| (001)0.59 | 0.153 (2.456) | 0.33 | |
| 4 | (001)0.87 | 0.153 (2.458) | 0.33 |
| 5 | (001)0.81 | 0.156 (2.495) | 0.40 |
| 6 | (001)0.99 | 0.158 (2.529) | 0.60 |
| 7 c | (001)0.41 | 0.160 (2.557) | 0.40 |
| (001)0.91 | 0.160 (2.557) | 0.40 | |
| 9 | (001)0.94 | 0.161 (2.582) | 0.40 |
| 10 | (001)1.00 | 0.164 (2.633) | 0.50 |
| 11 | (001)0.13 | 0.166 (2.657) | 0.17 |
| 12 | (001)0.06 | 0.179 (2.875) | 0.17 |
| 13 | (001)0.85 | 0.189 (3.032) | 0.00 |
| 14 | (001)0.15 | 0.192 (3.069) | 0.17 |
| Surface | Pre-Exponential Factor (ν) (s−1) | Kinetic Barrier Energy (eV) | Reaction Rate Constant (k) (s−1) |
|---|---|---|---|
| o-Fe7C3(001)0.85 | 1.24 × 1012 | 0.91 | 4.26 × 104 |
| o-Fe7C3(001)0.13 | 1.21 × 1012 | 1.48 | 8.66 × 10−1 |
| o-Fe7C3(001)0.20 | 1.30 × 1012 | 1.85 | 7.95 × 10−4 |
| o-Fe7C3(001)0.09 | 1.48 × 1012 | 1.74 | 7.72 × 10−3 |
| o-Fe7C3(001)0.99 | 1.22 × 1012 | 1.95 | 1.06 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chun, H.-J.; Kim, Y.T. Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations. Energies 2021, 14, 563. https://doi.org/10.3390/en14030563
Chun H-J, Kim YT. Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations. Energies. 2021; 14(3):563. https://doi.org/10.3390/en14030563
Chicago/Turabian StyleChun, Hee-Joon, and Yong Tae Kim. 2021. "Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations" Energies 14, no. 3: 563. https://doi.org/10.3390/en14030563
APA StyleChun, H.-J., & Kim, Y. T. (2021). Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations. Energies, 14(3), 563. https://doi.org/10.3390/en14030563